Hemodynamic alterations in cirrhosis and portal hypertension
Article information
Abstract
Portal hypertension (PHT) is associated with hemodynamic changes in intrahepatic, systemic, and portosystemic collateral circulation. Increased intrahepatic resistance and hyperdynamic circulatory alterations with expansion of collateral circulation play a central role in the pathogenesis of PHT. PHT is also characterized by changes in vascular structure, termed vascular remodeling, which is an adaptive response of the vessel wall that occurs in response to chronic changes in the environment such as shear stress. Angiogenesis, the formation of new blood vessels, also occurs with PHT related in particular to the expansion of portosystemic collateral circulation. The complementary processes of vasoreactivity, vascular remodeling, and angiogenesis represent important targets for the treatment of portal hypertension. Systemic and splanchnic vasodilatation can induce hyperdynamic circulation which is related with multi-organ failure such as hepatorenal syndrome and cirrhotic cadiomyopathy.
INTRODUCTION
Cirrhosis has been considered to be silent and static. However, we have recently recognized that cirrhosis is actually a tumultuous and dynamic disease. Cirrhosis is the final result of hepatic fibrosis and is reversible in the middle stages of development between fibrogenesis and fibrolysis. This disease leads to hemodynamic disorders that can have widespread impacts in the body according to the severity of the cirrhosis. Hemodynamic alterations including portal hypertension and hyperdynamic circulation are the main cause of morbidity and mortality in patients with cirrhosis.1-3 The pathophysiologic process of portal hypertension consists of three components: intrahepatic circulation, systemic (splanchnic) circulation, and collateral circulation. Additionally, continuous abnormalities in systemic circulation induce hyperdynamic circulation.4,5
Portal pressure is due to intrahepatic resistance and portal blood flow, and is defined as a function of flow and resistance across the hepatic vasculature (pressure=flow×resistance). Development of portal hypertension can be influenced by changes in resistance and flow in the hepatic vasculature. Increased resistance of portal blood flow in cirrhotic liver induces portal venous dilatation and congestion of portal venous flow, leading to elevated portal pressure. Subsequently, portosystemic collaterals develop to counterbalance the increased resistance in portal blood flow, and induce an increase in venous return to heart which results in increased portal venous inflow. This hyperdynamic splanchnic circulation contributes to the maintaince and aggravation of portal hypertension.4 Increased intrahepatic resistance results from both vasoconstriction and fibrosis. Vasoconstriction is a reversible and dynamic condition which contributes up to 25% of increased resistance (Fig. 1).5-7
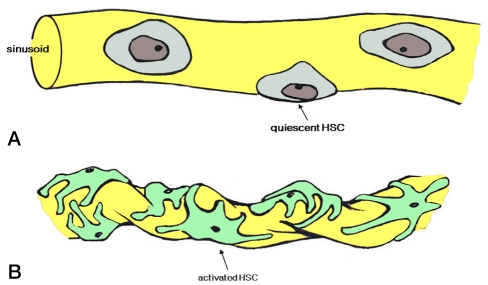
Hepatic stellate cell (HSC) activation. (A) In the quiescent state, HSCs do not contract. (B) In an activated state, the number and contractility of HSCs increase and induce changes in sinusoidal structure and intrahepatic resistance.
Vasoreactivity such as vasoconstriction in hepatic circulation and vasodilation in systemic circulation plays a major role in pathophysiology of portal hypertension.8 Recently, vascular structural changes including vascular remodeling and angiogenesis have been identified as additional important compensatory processes for maintaining and aggravating portal hypertension.9 Vascular remodeling is an adaptive response of the vessel wall that occurs in response to chronic changes in the environment such as shear stress.10 Angiogenesis promoted through both proliferation of endothelial and smooth muscle cells also occurs as response to increased pressure and flow. In this report, we review new concepts of pathophysiology and hemodynamic alterations associated with portal hypertension and cirrhosis.
Intrahepatic circulation
Vasoregulatory imbalances and increased intrahepatic resistance
Hepatic stellate cells (HSCs) play a central role in producing dynamic components of intrahepatic resistance by causing sinusoidal vasoconstriction through "contractile machinery" and relaxation in response to the interaction between sinusoidal endothelial cells (SECs) and HSCs; their paracrine effects are accomplished through endothelin-1 (ET-1) and nitric oxide (NO).11 Normally, ET-1 is secreted from SECs and acts on ETA receptors on HSCs leading to HSC contraction. Conversely, NO released from SECs by endothelial NO synthase (eNOS) induces relaxation of HSCs through the guanylate catalase pathway. Consequently, the balance between the ET-1 and NO accounts for the control of sinusoidal flow. However, in cirrhotic liver, the overproduction of ET-1 and increased susceptibility to autocrine ET-1 leading to activated HSCs result in increasing HSC contraction.12 In addition, multiple derangements in eNOS-derived NO generation by SECs contribute to impaired sinusoidal relaxation and increased intrahepatic resistance (endothelial dysfunction).13-15 Although eNOS protein levels appear to be unchanged, SECs show a prominent increase in the inhibitory protein caveolin binding to eNOS with concomitant decreased calmodulin binding, which may contribute to NOS dysfunction.13,16 Furthermore, recent studies have shown impaired phosphorylation and activation of eNOS mediated through alterations in G-protein coupled receptor signaling and defects in endogenous inhibitors of NOS, which suggest that multiple molecular defects likely contribute to a significant deficiency in hepatic NO production during cirrhosis.17 Sinusoidal vasoconstriction is due not only to diminished NO production by SECs, but also resistance of HSCs to NO due to defects in the guanylate cyclase signaling pathway.18,19 Animal experiments have demonstrated that activation of hepatic eNOS can improve portal hemodynamics in cirrhotic rat liver.8 Furthermore, a recent study evaluated the effects of simvastatin on intrahepatic vascular tone acting as an eNOS activator in humans.20 Patients who received simvastatin showed increased hepatic venous NO products and decreased hepatic vascular resistance without untoward systemic vascular effects.20
Sinusoidal Remodeling and Angiogenesis
HSC density and coverage of the sinusoidal lumen are increased in cirrhosis. The contractile nature and long cytoplasmic processes of HSCs encircling endothelial cells induce sinusoidal vessel constriction with increased vascular resistance termed "sinusoidal vascular remodeling". The characteristics of sinusoidal remodeling are distinct from process of fibrosis, collagen deposition of HSC.21,22 In this process, HSC motility and migration is absolutely required to promote enhanced coverage of HSCs around a SECs-lined sinusoid.
While Transforming growth factor-β (TGF-β) is largely recognized for its contribution to HSC-based collagen deposition, there is significant crosstalk between TGF-β and PDGF involved in HSCs motility. Indeed, these signals may converge at the level of c-abl tyrosine kinase.23,24 A number of signaling pathways mediate HSC recruitment to vessels in vascular remodeling and angiogenesis including PDGF, TGF-β, angiopoietins, and NO. Platelet derived growth factor (PDGF) is probably the most critical factor in the recruitment of pericytes to newly formed vessels.25 SECs also undergo substantive phenotypic changes in cirrhosis that likely contribute to changes in sinusoidal structure. Indeed, recent studies have identified a number of alterations in SEC phenotypes.26
Systemic and splanchnic circulation
Vasoregulatory imbalances in the splanchnic circulation
In contrast to diminished intrahepatic bioavailability of NO, splanchnic (and systemic) circulation shows a relative excess in regional NO generation.8 This increased production is largely endothelium-dependent,27 and is thought to be evidence of eNOS activation in splanchnic endothelium. Some studies have shown that eNOS activation by the angiogenic growth factor, vascular endothelial growth factor (VEGF), may be a primary factor in initial eNOS activation which demonstrates interesting links between vasodilating angiogenesis and vascular remodeling.28 Bacterial translocation during cirrhosis increases tumor necrosis factor-α (TNF-α) production which can also induce the increase of systemic NO production.29-31 Therefore, increased NO production in systemic and splanchnic circulation contributes to decreased systemic vascular resistance and resultant hyperdynamic circulation. This in turn results in sodium retension and ascites mediated by a reduction of effective circulating volume, stimulation of sympathetic system, an activation of the renin-angiotensin-aldosteron system, and an increase of antidiuretic hormone release (Fig. 2).

Pathogenesis of hyperdynamic circulation in cirrhosis and portal hypertension.
CO, cardiac output; eNOS, endothelial nitric oxide synthetase; NO, nitric oxide; HO, heme oxygenase; CM, carbon monoxide; TNF-α, tumor necrosis factor-α; RAA, rennin-angiotensin-aldosteron; SNS, sympathetic nerve system; ADH, anti-diuretic hormone; VEGF, vascular endothelial growth factor; HE, hepatic encephalopathy; CCM, cirrhotic cardiomyopathy; HRS, hepatorenal syndrome; HPS, hepatopulmonary syndrome.
Vascular remodeling of systemic vessels in portal hypertension
Vascular remodeling is a long-term adaptive response to chronic changes in blood flow. Chronic increases in flow with dilation of the vascular channel are implicated in endothelial-based signals that mediate restructuring of the vessel, thereby allowing for chronic increases in vessel diameter and capacity for high volume flow. This change has been demonstrated in peripheral vessels including experimental models of portal hypertension which may be related to activation of eNOS.10,32
Collateral circulation
Vasoregulatory imbalances in collateral circulation
The development of portosystemic shunts and collateral circulation such as esophageal and hemorrhoidal collateral vessels is a compensatory response to decompress the portal circulation and hypertension, but unfortunately contributes to significant morbidity and mortality. Vasodilation of pre-existing collateral vessels results in increased collateral blood flow and volume. The mechanism of collateral vessel regulation still remains unclear. The control of collateral circulation could be a key in managing complications of portal hypertension, therefore, experimental studies are performed.33
Angiogenesis and vascular remodeling in collateral circulation
In addition to vasodilatation, the collateral circulatory bed develops through angiogenesis. Angiogenesis occurs through the proliferation of endothelial and smooth muscle cells in addition to vasculogenesis. Vasculogenesis refers to the recruitment of endothelial progenitor cells for the de novo synthesis of vessels.34 Angiogenesis and vasculogenesis are also influenced by NO and highly dependent on VEGF as the growth factor exerting pleiotropic effects to promote new vessel formation.35,36 Indeed, VEGF promotes vasodilation, vascular remodeling, and angiogenesis in part through NO-dependent or independent mechanisms. In animal models, neutralizing antibodies inhibited portosystemic shunting by blocking VEGF receptor 2, which further highlights the importance of VEGF and NO for increased portosystemic collateralization in portal hypertension.37 In addition, multikinase inhibitors such as sorafenib result not only in decreases of portosystemic shunts and improvement of portal hypertension but also inactivation of HSCs. This is under active investigation,37-39 however, more studies are needed for clinical application.
Hyperdynamic circulation
The hyperdynamic circulation is characterized by increased cardiac output and heart rate, and decreased systemic vascular resistance with low arterial blood pressure in cirrhotic patients.40-43 These hemodynamic alterations are initiated by systemic and splanchnic vasodilatation, and eventually lead to abnormalities of the cardiovascular system and several regional vascular beds including ones involved in hepatic, splanchnic, renal, pulmonary, skeletal muscle and cerebral circulation.5
Clinical features and pathogenesis
Hyperdynamic circulation is clinically presents with tachycardia, hypotension, and bounding pulses. Although hyperdynamic circulation per se is not distressing to the patient, this phenomenon is clinically relevant due to its propensity to aggravate or precipitate some of the complications associated with portal hypertension. Severity of hyperdynamic circulation correlates with advancing liver failure with patients with end-stage liver failure generally showing the greatest extent of peripheral vasodilatation and increased cardiac output.41-44 Thus, virtually all patients with decompensated cirrhosis show evidence of hyperdynamic circulation. However, the presence of portal hypertension, rather than liver failure, is essential for the development of hyperdynamic circulation. Since the gut and liver receive a third of the entire cardiac output, hyperdynamic circulation directly or indirectly contributes to two of the most troublesome complications of cirrhosis: ascites and variceal bleeding. In concert with the increased total cardiac output, mesenteric blood flow also increases.45,46 Moreover, studies in both humans and animal models of cirrhosis or portal hypertension confirm that mesenteric hyperemia is due not only to a passive increase in blood flow as part of the increased cardiac output, but also to mesenteric vasodilatation. In other words, the percentage of overall cardiac output perfusing the mesenteric organs also increases.42,43,45,46 Recently, bacterial infection has been recognized as a risk factor for precipitating variceal bleeding.47 The underlying mechanism of this curious observation remains unknown, but it has been suggested that humoral substances released during the course of sepsis, including endotoxins and cytokines such as TNF-α, intensify the hyperdynamic circulation and thus increase blood flow through varices. The exact pathogenic mechanisms leading to hyperdynamic circulation remain to be definitively determined. Several factors to date have been hypothesized to be involved, including humoral substances, central neural activation, tissue hypoxia, and hypervolemia.48
Multi-organ involvement
Heart
Cirrhotic cardiomyopathy was first described in the late 1960s although it was mistakenly attributed to latent or subclinical alcoholic cardiomyopathy for many years.49-51 Despite an increased baseline cardiac output, cirrhotic patients have a suboptimal ventricular response to stress. These individuals show blunted systolic and diastolic contractile responses to stress in conjunction with evidence of ventricular hypertrophy or chamber dilatation, and electrophysiological abnormalities including prolonged QT intervals. The pathogenesis of this syndrome is multifactorial and includes diminished β-adrenergic receptor signal transduction,2,52-55 cardiomyocyte cellular plasma membrane dysfunction, and increased activity or levels of cardio-depressant substances such as cytokines, endogenous cannabinoids, and nitric oxide.51 Although cirrhotic cardiomyopathy is usually clinically mild or silent, overt heart failure can be precipitated by stress from liver transplantation or transjugular intrahepatic portosystemic shunt insertion. Recent studies suggest that the presence of cirrhotic cardiomyopathy may contribute to the pathogenesis of hepatorenal syndrome precipitated by spontaneous bacterial peritonitis,56 acute heart failure after insertion of transjugular intrahepatic portosystemic shunts,57,58 and increased cardiovascular-associated morbidity and mortality after liver transplantation.59
The Kidney
Renal vasoconstriction is characteristic in kidney with splanchnic vasodilation and hyperdynamic circulation, and may be responsible for the development of hepatorenal syndrome. Renal vasoconstriction develops as a consequence of effective hypovolemia and ensuing neurohumoral activation.60 This provides the rationale for treating hepatorenal syndrome with albumin infusion and vasoconstrictors (terlipressin, norepinephrine, or midodrine).61
The Lung and Brain
Vasodilatation in the lung leads to ventilation perfusion mismatch and even arterio-venous shunts in the pulmonary circulation; these result in hepatopulmonary syndrome, characterized by marked hypoxemia.62,63 In some cases, this may evolve into the opposite situation with markedly increased pulmonary vascular resistance seen in portopulmonary hypertension.64 This is thought to develop through endothelial dysfunction and vascular remodeling of the pulmonary circulation.65 Changes in cerebral blood flow and vascular reactivity associated with portal hypertension are considered to contribute and facilitate some of the brain abnormalities of hepatic encephalopathy.
CONCLUSIONS
Portal hypertension is associated with vascular alterations in intrahepatic and systemic circulation. Extensive research has improved our understanding of the pathogenic mechanisms underlying hemodynamic derangement, allowing the development of novel treatment modalities. Future studies should focus on pharmacologic and genetic approaches to modulate vascular biologic systems to ameliorate complications and symptoms relating to hemodynamic alterations in patients with cirrhosis and portal hypertension.
Acknowledgements
This work was supported by a grant from Ministry for Health, Welfare and Family Affairs, Republic of Korea (no. A050021).
Abbreviations
HSCs
hepatic stellate cells
SECs
sinusoidal endothelial cells
ET-1
endothelin-1
No
nitric oxide
eNOS
endothelial NO synthase
CO
cardiac output
HO
heme oxygenase
CM
carbon monoxide
TNF-α
tumor necrosis factor-α
RAA
rennin-angiotensin-aldosteron
SNS
sympathetic nerve system
VEGF
vascular endothelial growth factor
HE
hepatic encephalopathy
CCM
cirrhotic cardiomyopathy
HRS
hepatorenal syndrome
HPS
hepatopulmonary syndrome
TGF-β
transforming growth factor-β
PDGF
platelet derived growth factor