INTRODUCTION
Primary biliary cirrhosis (PBC) is a slowly progressive cholestatic liver disease of autoimmune etiology.1 PBC is characterized by presence of antimichondrial antibody (AMA), histologic findings of portal inflammation and immune-mediated destruction of the intrahepatic bile ducts. It mainly affects middle-aged women. PBC is most prevalent in northern Europe. The prevalence of PBC differs considerably in different geographic regions, ranging from 40 to 400 per million.1 The prevalence of PBC in Japan is about from 27 to 54 per million.2 The prevalence of PBC in Korea has not been investigated, but PBC is designated as one of rare disorders by Korean government. The clinical characteristics of PBC in Korea are similar with those in regions where PBC are prevalent.3 The manifestations and prognosis are various in different patients. Diagnosis in earlier stage and treatment with ursodeoxycholic acid (UDCA) have improved the prognosis in patients with PBC over the past two decades. This article reviews an overview of the updated knowledge on the diagnosis and treatment of PBC.
NATURAL HISTORY
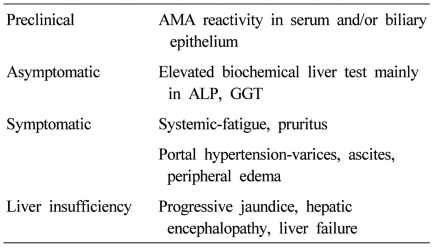
PBC progresses insidiously through the clinical phases: preclinical, asymptomatic, symptomatic, and liver insufficiency (Table 1).4 The preclinical phase is characterized by AMA reactivity with no symptom and normal biochemical liver tests. Then patients develop biochemical abnormalities but remain asymptomatic. The median time to progression from preclinical to asymptomatic phase was 5.6 years (range, 1-20 years).5 Asymptomatic phase is followed by the development of symptoms, usually fatigue and pruritus, and later varices, edema, or ascites in most untreated patients within 2 to 4 years.6 Liver insufficiency is characterized by accelerated jaundice, and the prognosis is poor.7 Mean survival in patients with bilirubin level of 2.0 mg/dL is 4 years, and that in patients with bilirubin level of 6.0 mg/dL is 2 years.
The prognosis of patients with PBC has improved significantly over the past 2 decades because more patients are being diagnosed earlier in the disease process8 and being treated with UDCA. UDCA therapy significantly delayed histologic progression,9 decreased the development of esophageal varices,10 and increased the survival in patients with PBC.11-13 The survival rate of patients with early stage (stage 1 or 2 disease) who were treated with UDCA for a mean of eight years was similar to that of a healthy control population.14
CLININCAL MANIFESTATIONS
PBC is now diagnosed earlier in its clinical course owing to easy access to biochemical tests and widespread use of the specific AMA assay. More than 50% of patients are asymptomatic at presentation.3,15-17 Sixty percents of patients were asymptomatic at diagnosis also in Korea.3 The most common symptoms in PBC patients at diagnosis are fatigue and pruritus. Fatigue has been reported in up to 78 percents of patients,18-20 and does not appear to correlate with disease severity, histologic stage, or duration, and may impair the quality of life.20 The etiology of fatigue is unknown, but may be related to autonomic dysfunction.21 Pruritus, which occurs in 20 to 70 percent of patients, can be the most distressing symptom.22 The onset of pruritus usually precedes the onset of jaundice by months to years. The pruritus can be local or diffuse. It is usually worse at night and is often exacerbated by contact with wool, other fabrics, or heat. Its cause is unknown, but endogenous opioids may have a role. Unexplained discomfort in the right upper quadrant occurs in approximately 10 percent of patients.23 Other common findings in primary biliary cirrhosis include hyperlipidemia, hypothyroidism, osteopenia, and coexisting autoimmune diseases such as Sjögren's syndrome and scleroderma.24 Portal hypertension does not usually occur until later in the course of the disease. Malabsorption, deficiencies of fat-soluble vitamins, and steatorrhea are uncommon except in advanced disease. Rarely, patients present with ascites, hepatic encephalopathy, or hemorrhage from esophageal varices.25 The incidence of hepatocellular carcinoma is elevated among patients with long-standing advanced disease.26 Other diseases associated with primary biliary cirrhosis include interstitial pneumonitis, celiac disease, sarcoidosis, renal tubular acidosis, hemolytic anemia, and autoimmune thrombocytopenia.
DIAGNOSIS
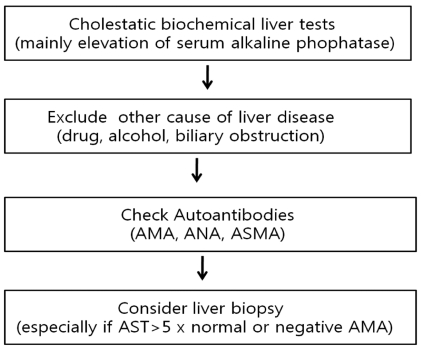
The diagnosis of PBC should be suspected in the setting of chronic cholestasis after exclusion of other causes. The diagnosis is based on the following findings; 1) biochemical evidence of cholestasis with elevated alkaline phophatase (ALP) activity and/or gamma glutamyl transpeptidase (GGT), 2) presence of antimitochondiral antibody (AMA), and 3) histologic evidence of nonsuppurative cholangitis and destruction of interlobular bile ducts (Fig. 1). Patients are diagnosed as probable PBC if two of these three features are present after exclusion of biliary obstruction.27
Liver biochemical tests
The biochemical hallmarks of PBC are elevated serum ALP and GGT. Mild elevation of serum aminotransferases and increased level of immunoglobulins (mainly IgM) is commonly observed16 while some patients with PBC may have high aminotransferase activities with hypergammaglobulinemia. The changes in biochemical tests reflect in part the severity of histology28,29 and the improvement of biochemical tests after UDCA administration is a strong predictor of long-term prognosis.30-33 In patients without cirrhosis, the degree of ALP elevation is related to the severity of ductopenia and inflammation on liver histology. The increase in aminotransferase and IgG levels reflects the degree of periportal and lobular necroinflammation. The level of serum bilirubin reflect the severity of ductopenia and biliary piecemeal necrosis.29 Hyperbilirubinemia, hypergammaglobulinemia, hypoalbuminemia, and thrombocytopenia are indicators of the development of liver cirrhosis and portal hypertension. As in other chronic cholestatic disease, serum cholesterol levels often elevated.28,29
Autoantibodies
AMA was described in a patient with PBC in 1965 for the first time,34 and has been regarded as a hallmark of PBC. Serum AMA is highly specific for the diagnosis of PBC and detected in nearly 95% of patients with PBC, while it is detected in normal population in about 1%.1 When AMA is detected in asymptomatic subjects with normal biochemical tests, PBC is already present histologically in 40% of cases,35 and in the remaining patients it is likely to develop in succeeding years.5,16,36,37 The antigenic target of AMA is the E2 subunits of 2-oxo acid dehydrogenase complexes, in particular the pyruvate dehydrogenase complex (PDC)-E2.38 AMA titer may differ by more than 200-fold among patients who have PBC, but in the single patient it remains stable over the years, and has no prognostic value in PBC.39 Therefore, presence of AMA itself rather than its titer is important for the diagnosis. The measurement of serum AMA is typically based on the immunofluorescent techniques (the criteria of positivity, above 1:40), however, with recognition of antigenic determinants, enzyme-linked immunosorbent assays (ELISA) or western blotting assays have been developed. Each assay uses different epitopes, so that interpretation of AMA result should be considered the different sensitivity and specificity of the specific assay. Moreover, 5-10% of PBC patients did not show AMA positivity in their sera, so that liver biopsy is required for the suspicious cases. However, the comparison of AMA-positive PBC and AMA-negative PBC did not show significant differences in terms of clinical features, treatment response or prognosis.40 Antinuclear antibodies (ANA) and anti-smooth muscle antibody (ASMA) are found in about half of PBC patients. ANAs such as anti-gp210 and possibly anti-p62 are detected in some of patients with PBC and may be associated with aggressive disease and poor prognosis.41-43
Histology
Histology of PBC is characterized by chronic, nonsuppurative cholangitis that affects interlobular and septal bile ducts. The term "florid duct lesion" is often used when focal lesions show intense inflammatory changes and necrosis around the small bile ducts. The inflammatory infiltrates are comprised of plasma cells, macrophages, polymorphonuclear cells (especially eosinophils) and sometimes epithelioid granulomas.1 The size of the specimen is important and at least 10-15 portal tracts should be present to adequately evaluate cholangitis and ductopenia.
Histologic lesions are classically divided into four stages. Stage I is characterized by portal inflammation with or without florid duct lesion and the inflammation is confined to the portal triads. Stage II is a progression of periportal lesions to involvement of the hepatic parenchyma, which is termed as interface hepatitis. Stage III is characterized by distortion of the hepatic architecture with numerous fibrous septa. Stage IV is defined as cirrhosis with the existence of regenerative nodules.44
The role of liver biopsy for the diagnosis of PBC is limited, when the biochemical liver tests and AMA results are compatible to PBC. However, the information on the stage of PBC and the exclusion of the possibility of overlap syndrome such as autoimmune hepatitis can be obtained from the liver biopsy results. For AMA-negative patients, liver biopsy is mandatory for the diagnosis of PBC. Therefore, balanced decision should be made to do or not to do liver biopsy for suspicious PBC patients after consideration of benefit and risk or cost related to the invasive procedure.
TREATMENT
Ursodeoxycholic Acid (UDCA)
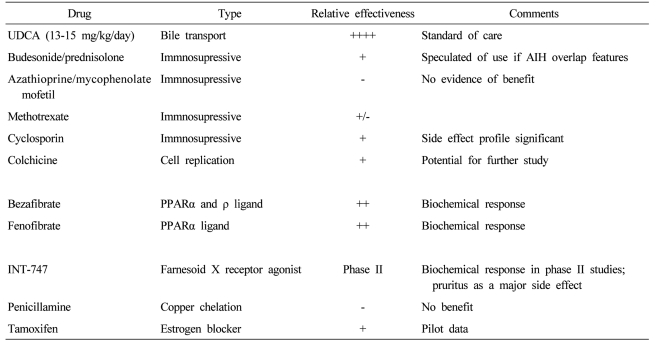
UDCA, the 7-beta epimer of chenodeoxycholic acid, comprised 2% of human bile acid and has several interrelated functions including direct choleretic, anti-inflammatory, and antiapoptotic properties.45 UDCA in a dose of 13-15 mg/kg /day is the only drug approved for PBC treatment by USA FDA (Table 2).27 It decreases serum levels of bilirubin, ALP, aminotransferase, cholesterol and IgM,46,47 and improves liver histology.12,46 A combined analysis of the three largest clinical trials shows that UDCA prolongs survival free of liver transplantation.48 The adequate dosing of UDCA is important. A dose of 13-15 mg/kg/day was superior to either a lower dose of 5-7 mg/kg/day or a higher dose of 23-25 mg/kg/day in biochemical responses and costs.49 A complete response occurs in about 30% of patients treated with PBC, which was defined by normalized biochemical tests and stabilized or improved liver histology.50,51 Serum ALP levels at 6 months after UDCA therapy can be helpful in predicting response to UDCA.30,32 The life expectancy of patients showing the complete response during treatment with UDCA was similar to that of age- and sex-matched healthy controls for up to 20 years.32 However, the disease progresses in many patients who do not show the complete response during UDCA therapy, for whom additional medical treatment is definitely required.
Other drugs than UDCA
Other drugs for the treatment of PBC have been studied for the past decade as single agents or adjuvant medications. None of these drugs have been found beneficial as single agents, in which colchicines,52,53 methotraxate,54 penicillamine,55 cyclosporine,56 corticosteroid,57 azathioprine,58 mycophenolate mofetil were included.59 Many of these have been used in combination with UDCA to see if further improvement in liver disease can be achieved. Budesonide had been reported to improve liver histology and the biochemical tests of liver function when used with UDCA, but it may worsen osteopenia.60 However, studies were of too short treatment duration to show convincingly whether budesonide will improve survival or not. The additions of colchicines,61 methotraxate62 and silymarin63 to UDCA had no additional benefits compared with UDCA alone.
Bezafibrate and fenofibrate (fibric acid derivatives used to treat hypertriglyceridemia) improved liver biochemical tests in pilot studies.64,65 The proposed mechanism of action of fibric acid derivatives in treatment of PBC involves the regulation of expression of immunomodulatory proteins and lipids,66,67 downregulation of cholesterol 7 alpha-hydroxylase, an enzyme involved in the synthesis of bile acids68 and decrease of multi-drug resistance (MDR) gene through the activation of peroxisome proliferator-activated receptor alpha.69 Farnesoid X receptor (FXR) is a bile acid-activated nuclear receptor highly expressed in both the liver and gastrointestinal tract. It has a regulatory role in bile and cholesterol metabolism, and FXR agonists such as INT-747 may hold promise for the new therapeutic option in UDCA-refractory PBC.70 Modification of UDCA (nor-UDCA) with more potent choleretic property than UDCA improved liver biochemistry and histology in MDR2 knockout mouse, an animal model for slcerosing cholangitis, which suggests that translational studies in human are required.71 Tamoxifen decreased alkaline phosphatase levels in two women who were taking it after surgery for breast cancer.72 Although atorvastatin with many antiinflammatory properties was commonly used for control of hypercholesterolemia in PBC, it was not effective for PBC itself.73
Liver transplantation
Transplantation is the only effective treatment for those with decompensated cirrhosis or liver failure.74 The outcome of liver transplantation in patients with PBC is more favorable than for other liver diseases. The survival rates at one and five years are 92 percent and 85 percent, respectively. About 20-25% of patients with PBC who undergo transplantation have recurrent disease over 10 years. Fortunately, recurrent PBC does not affect patient or graft survival.75
CONCLUSION
The diagnosis of PBC should be suspected in subjects with chronic cholestasis, mainly elevation of ALP after exclusion of other causes of hepatobiliary disease. The diagnosis of PBC is largely confirmed with tests for AMA. AMA is highly specific for the diagnosis of PBC and positive in nearly 95% of patients. Liver biopsy can be used especially for further evaluation in subjects with negative tests for AMA and suspicious overlap syndrome. UDCA in a dose of 13-15 mg/kg /day is the standard therapy for PBC treatment, however, about 40% of the patients do not respond to UDCA. Therefore, further efforts to develop new drugs and clinical trial should be warranted.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print