Recent research trends and updates on nonalcoholic fatty liver disease
Article information

Abstract
Nonalcoholic fatty liver disease (NAFLD), together with metabolic syndrome and obesity, has shown a rapid increase in prevalence worldwide and is emerging as a major cause of chronic liver disease and liver transplantation. Among the various phenotypes of NAFLD, nonalcoholic steatohepatitis (NASH) is highly likely to progress to development of end-stage liver disease and cardiometabolic disease, resulting in liver-related and non-liver–related mortality. Nonetheless, there is no standardized pharmacotherapy against NASH and many drugs are under development in ongoing clinical trials. To develop a successful anti-NASH drug, it is necessary to select an appropriate target population and treatment outcomes depending on whether the mode of action is anti-metabolic, anti-inflammatory or anti-fibrotic. Recently, innovative surrogate markers have been investigated to replace hard outcomes such as liver histology and mortality and reduce the clinical trial duration. Currently, several drugs with fast track designation are being tested in phase III clinical trials, and many other drugs have moved into phase II clinical trials. Both lean NAFLD and typical obese NAFLD have been extensively studied and genetic variants such as PNPLA3 and TM6SF2 have been identified as significant risk factors for lean NAFLD. In the near future, noninvasive biomarkers and effective targeted therapies for NASH and associated fibrosis are required to develop precision medicine and tailored therapy according to various phenotypes of NAFLD.
INTRODUCTION
Although the prevalence of nonalcoholic fatty liver disease (NAFLD) in Korea has recently increased, no drug has been recognized as standard pharmacotherapy and many drugs remain in the clinical trial stage [1]. Pharmacotherapy is typically not indicated for patients with simple steatosis but is focused on histological improvement of nonalcoholic steatohepatitis (NASH) and fibrosis [2]. For approval of anti-NASH drugs, key endpoints required for a clinical trial design must be met and standardized internationally. Recently, complete resolution of NASH without worsening of liver fibrosis has been considered an optimal surrogate endpoint in clinical trials for NASH [3]. Approximately 130 clinical trials are underway in relation to NASH treatment, with only five drugs undergoing phase III, while the remaining drugs remain in phase I or II [4]. In this review, we summarize definitions and key endpoints that are considered to be important when conducting clinical trials for NASH, and introduce the new drugs that are drawing attention in each phase. In addition, we will briefly describe non-obese NAFLD, which is emerging as a new phenotype of NAFLD.
DEFINITIONS OF NAFLD/NASH IN CLINICAL TRIALS
NAFLD and NASH
NAFLD is defined as the presence of ≥ 5% macrovesicular steatosis [3]. According to adequate specimen criteria, > 10 portal tracts should be included in ≥ 2 cm of tissue to allow appropriate evaluation. NASH is diagnosed based on an overall pattern of histological hepatic injury consisting of macrovesicular steatosis, inflammation, or hepatocellular ballooning [2,5-7]. In the case of definite steatohepatitis , all three conditions should be fulfilled [3,8]. In the case of borderline steatohepatitis , ballooning and Mallory-Denk body are absent or atypical [3,5]. The common definitions of the histological spectrum of NAFLD are listed in Table 1 [3].
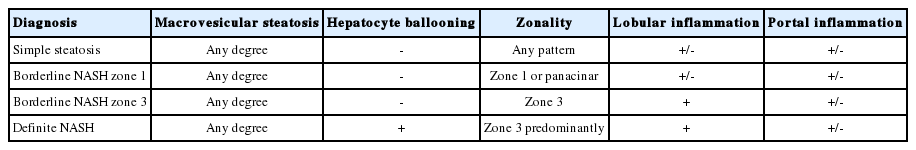
Diagnostic criteria of the histological lesions in NAFLD/NASH
Disease activity
It is advisable to use the NAFLD activity score (NAS), which is a sum of three histological components: steatosis (0–3), lobular inflammation (0–3), and hepatocellular ballooning (0–2) [3,5]. It is generally accepted that NAS ≥ 4 points is likely to indicate steatohepatitis. However, evidence for an association between the NAS and long-term outcomes is lacking and a lucid working definition of NASH is needed to define inclusion criteria of clinical trials. The NAS is commonly used to identify the severity of NAFLD or to reflect changes in histological findings, rather than to diagnose NASH [9].
Disease stage
It is recommended that the Nonalcoholic Steatohepatitis Clinical Research Network (NASH CRN) fibrosis stage be used to identify the disease stage as the most widely validated method [3,5]. The NASH CRN staging system basically mimics the METAVIR system for determining fibrosis severity except for the subclassification of stage 1 into substages a-c depending on the location of collagen deposition.
ENDPOINTS OF CLINICAL TRIALS FOR NAFLD/NASH
Clinical outcomes
Common endpoints of clinical trials in NAFLD/NASH are summarized in Table 2. Clinical outcomes considered in NASH clinical trials include progression to cirrhosis, progression to decompensation (ascites, encephalopathy, and variceal hemorrhage), all-cause mortality, and liver-related mortality [10]. However, progression from NASH to cirrhosis takes almost 30 years and research resources are limited [10]. In addition, only a minority of patients with NASH progress to cirrhosis or morbidity/mortality compared to those with viral hepatitis, and NASH-associated hepatocellular carcinoma (HCC) occasionally arises from non-cirrhotic but steatotic liver [11-14]. Therefore, fibrosis progression rather than traditional hard endpoints may be alternatively evaluated and monitored using transient elastography (TE) and magnetic resonance elastography (MRE) [15-18]. Liquid biomarkers such as pro-C3, fibrosis-4 (FIB-4) index, NAFLD fibrosis score (NFS), and enhanced liver fibrosis (ELF) test can also be used as surrogate endpoints [19-22]. Decompensation can be indirectly assessed by Child-Pugh score, the model for end-stage liver disease (MELD) score, and hepatic venous pressure gradient (HVPG). Particularly, in the case of Child-Pugh score, a change of ≥ 2 points is regarded as a meaningful change [3,5,10,23]. However, these markers have inherent limitations. First, the above surrogate endpoints are not specific for NASH, and several Child-Pugh score components are subjective and may create information bias. Second, due to the invasive nature of HVPG and the need for serial monitoring, its applicability beyond clinical trials is debatable.
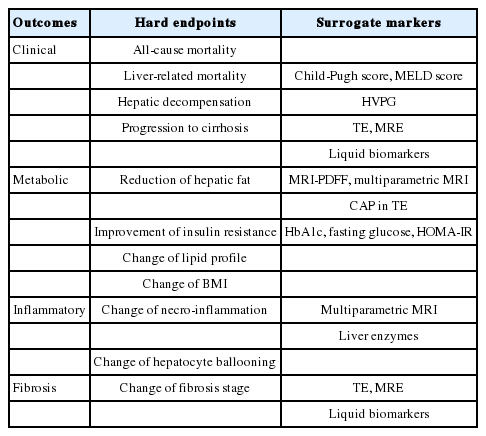
Common endpoints of clinical trials in NAFLD/NASH
Metabolic outcomes
Important indicators reflecting metabolic outcomes include changes in liver fat content, insulin resistance, lipid profiles, and body mass index (BMI) [23]. Among these, paired biopsy is the gold standard for evaluation of improvement in liver fat content. However, liver biopsy is invasive and semiquantitative and patient compliance is relatively low, resulting in a higher dropout rate. Recently, the techniques of magnetic resonance imaging–derived proton density fat fraction (MRI-PDFF) and multiparametric MRI, which can measure liver fat content noninvasively, have been developed and validated [24-26]. These methods are not only noninvasive but have the additional advantage of measuring liver fat content across the whole liver compared to liver biopsy [27]; however, they are not commonly used in real practice due to their high cost. The controlled attenuation parameter (CAP) of TE may be another option although it requires further validation [16,23,28].
Insulin resistance is one of the main pathophysiologic mechanisms of NAFLD and also an important metabolic treatment outcome of pharmacotherapy against NASH [2,29,30]. The hyperinsulinemic-euglycemic clamp technique is the gold standard to measure insulin sensitivity but because of its cumbersome process and requirement for hospitalization, homeostasis model assessment for insulin resistance (HOMA-IR), fasting blood glucose, and glycosylated hemoglobin (HbA1c) are more frequently used as surrogate markers of metabolic outcomes [31-35].
Inflammatory outcomes
An important indicator of inflammatory outcomes is the change in necro-inflammation or hepatocyte ballooning as assessed by histological examination [23]. To date, complete resolution of NASH without worsening of fibrosis is the most commonly targeted outcome for proving efficacy of drugs for NASH, although there are several limitations pertaining to that goal. For example, reversal of NASH has not been shown to reduce overall or liver-related mortality, and to reduce inflammation severity gradually as fibrosis progresses.
The NAS is commonly used for evaluation of inflammatory outcomes. Although the NAS has not been shown to predict longterm prognosis or mortality, it is the most validated histological scoring system to date. In order for a drug to be recognized as an effective treatment, histological resolution of steatohepatitis without worsening of fibrosis should be achieved [3,4]. Recently, noninvasive multiparametric MRI has been used to evaluate inflammation, and liver enzymes, such as aspartate aminotransferase (AST) and alanine aminotransferase (ALT), are also used as biochemical surrogate markers [8,36-38]. When ALT is used as a surrogate marker, a decrement by >30-40% from the baseline is considered a robust response [39-42].
Fibrosis outcomes
In general, fibrosis is considered to have improved when a reduction by at least one stage has been achieved in terms of histological examination [3,5]. In phase IIa clinical trials, serologic tests can be used as surrogate endpoints to predict the severity of fibrosis instead of liver biopsy [3,10,23,43-45]. The most verified tests to predict fibrosis are NFS, FIB-4, ELF, and FibroTest® (Biopredictive, Paris, France), and these tests have also been studied in relation to overall and liver-related mortality [19-22].
Among the imaging modalities to predict fibrosis, TE is the most widely used in recent clinical trials and has been well documented in many studies [46]. However, TE has lower diagnostic accuracy for advanced fibrosis (F3) than for cirrhosis (F4), and may be unfeasible in cases of morbid obesity [47,48]. Therefore, special attention should be paid when interpreting TE results in clinical trials for NASH.
CURRENT PHA RMAC OTHERAPY UNDER DEVELOPMENT IN PHASE II/III TRIALS
Next, we will discuss the study design and regulatory pathway required for phase II and III clinical trials. The timeline, endpoints, and surrogate markers for each trial phase are summarized in Table 3 [4]. A phase IIa trial is a proof-of-concept study to confirm the mechanism of action of the investigational drug, and histological examination is not mandatory in this phase. In addition, since the treatment duration of a phase IIa trial is relatively short, anti-inflammatory or anti-metabolic efficacy rather than anti-fibrotic efficacy is often evaluated. It is important to select appropriate surrogate markers to evaluate treatment outcomes because ethical issues may be raised if paired biopsy is performed in a short time span. However, in phase IIb and III trials, paired biopsy is indispensable for testing the safety and tolerability of new drugs. Phase III clinical trials are classified as registrational trials for the purpose of marketing [4,23]. Generally, the surrogate marker is first used to receive conditional approval, and subsequent definite approval can be obtained later when the hard endpoint would be satisfied [10]. For conditional approval, one of the following two surrogates must be satisfied: (i) resolution of NASH without worsening of fibrosis and (ii) a reduction in fibrosis by one or more stage without worsening of NASH [4]. It is not necessary to satisfy both conditions simultaneously, but the choice of proper endpoints may be dependent on whether the mechanism of the drug is anti-inflammatory, anti-metabolic, or anti-fibrotic. Below, we briefly introduce the representative drugs under development in each phase.
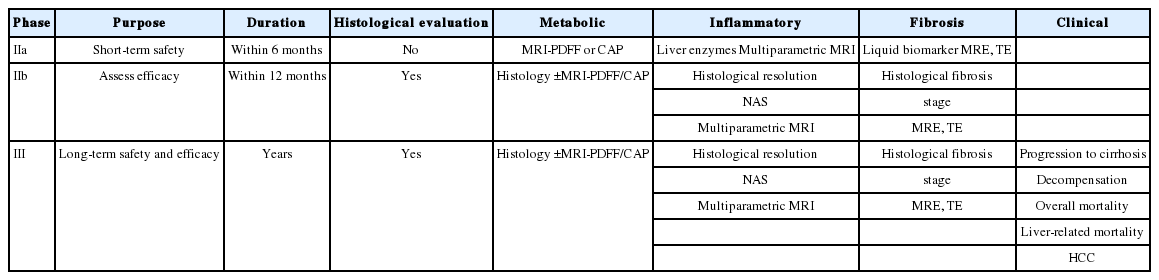
Characteristics of phase-specific clinical trial design
Phase IIa
Recombinant fibroblast growth factor 19 (FGF19) (NGM282)
The proportion of patients with liver fat reduction ≥ 5% on MRI-PDFF was significantly higher in the treatment group than in the placebo group (79% vs. 7%) when 82 patients with biopsyproven NASH were treated for 12 weeks [49]. The ALT normalization rate was 35% and 2% in the treatment group and the placebo group, respectively.
FGF21 agonist (BMS-986026)
The reduction rate of hepatic fat fraction on MRI-PDFF was -8.8% in the treatment group and -1.3% in the placebo group when 74 patients with biopsy-proven NASH and BMI ≥ 25 were treated for 16 weeks [4]. The severity of fibrosis on MRE was improved by 36% in the treatment group and by 15% in the placebo group, and the metabolic parameters, adiponectin level, and lipid profiles were ameliorated in the treatment group.
Glucagon like peptide 1 (GLP-1) agonist (Liraglutide and Semaglutide)
Forty-five biopsy-proven NASH patients with overweight were treated with either 1.8 mg of liraglutide or placebo for 48 weeks. Based on an improved ballooning score, the resolution rate of NASH was higher in the liraglutide group vs. the placebo group. However, liraglutide treatment failed to show improvement of fibrosis [50]. Currently, semaglutide is under clinical evaluation as a longer-acting alternative to liraglutide.
Acetyl-CoA carboxylase (ACC) inhibitor (GS-0976, PF-05221304)
In 12 patients with biopsy-proven NASH treated with 20 mg of GS-0976 for 12 weeks, liver fat contents assessed by MRI-PDFF and liver stiffness on MRE were decreased compared to the placebo group [4].
Phase IIb
Stearoyl CoA desaturase 1 (SCD1) inhibitor (Aramchol)
In the phase IIa trial, 60 patients with biopsy-confirmed NAFLD and 6 patients with biopsy-proven NASH were treated with 300 mg aramchol for 12 weeks. The treatment group showed a significant decrease in liver fat content compared to the placebo group (12.6% vs. 6.4%) [51]. Based on this finding, a phase IIb clinical trial targeting diabetes, overweight, and biopsy-confirmed NASH patients is underway.
Pan-caspase inhibitor (Emricasan)
In the phase IIa trial, HVPG decreased in 17.2% of patients with mean HVPG of 12 mmHg at baseline and ALT was significantly decreased in the emricasan group [4]. Based on these data, several phase IIb trials are currently underway in NASH patients with either compensated cirrhosis or decompensated cirrhosis.
Galectin-3 inhibitor (GR-MD-02)
In the phase IIa trial, 30 patients with biopsy-proven NASH and advanced fibrosis were treated for 16 weeks but did not meet the primary endpoint, which was fibrosis improvement as assessed by MRE [4]. The phase IIb clinical trial for patients with compensated NASH cirrhosis and portal hypertension is currently ongoing with extended treatment duration of 52 weeks. For cirrhosis, liver fibrosis assessed by MRI would be evaluated, and for portal hypertension, HVPG would be measured after 1 year of treatment.
Phase III
Farnesoid X receptor (FXR) agonist (Obeticholic acid, OCA)
In the phase IIb trial, 283 biopsy-proven NASH patients without cirrhosis were treated with OCA vs. placebo for 72 weeks [52]. The proportion of patients with histological improvement by >2 points of the NAS was 45% in the OCA group but only 21% in the placebo group. However, the rates of complete resolution of NASH and fibrosis improvement were not different between the two groups. To obtain Food Drug Administration approval, the phase III clinical trial is currently underway in 2,000 patients with NASH and fibrosis, and the study endpoints include histological improvement at 18 months, mortality at 6 years, and liver-related events. The study will evaluate not only the improvement of NASH but also the adverse events of OCA, such as pruritis and change of lipid profiles, that were observed in the phase II trials [52].
Peroxisome proliferator-activated receptor alpha/delta (PPAR-α/δ) agonist (Elafibranor)
A total of 276 non-cirrhotic patients with NASH were included in the phase IIb trial [38]. The proportion of patients with resolution of NASH was 23% in the treatment group and 17% in the placebo group, which was not significantly different. However, when the definition of NASH resolution was based on more stringent criteria recommended by regulatory authorities (disappearance of ballooning and none or mild persistence of lobular inflammation), a significant histological improvement was found in the elafibranor 120 mg group with good stability results. Currently, the phase III trial of 2,000 patients with NASH is underway for further evaluation. In addition to histological assessment, the effects of elafibranor on drug-related mortality, liver-related complications, and cardiovascular disease would be evaluated at 72 weeks.
Chemokine receptor 2 and 5 (CCR2/5) antagonist (Cenicriviroc, CVC)
In the phase IIb clinical trial, patients with NASH and fibrosis were treated with CVC for 2 years and histological changes were evaluated [53]. In the first year of the interim analysis, the primary outcome was not satisfactory but the anti-fibrotic effect of CVC in patients with severe NASH was prominent and the phase III clinical trial is currently ongoing.
Apoptosis-signal regulating kinase 1 (ASK1) inhibitor (Selonsertib)
In the recent randomized, open-label, phase II trial, NASH patients with moderate to progressive hepatic fibrosis were treated with either selonsertib alone or the combination of selonsertib and simtuzumab for 24 weeks [4]. The selonsertib alone group showed a significant improvement in liver fibrosis compared to the simtuzumab alone group. Large-scale phase III trials have been started in NASH patients with advanced liver fibrosis or cirrhosis.
APPROACH TO NON-OBESE NAFLD AS A NEW PHENOTYPE OF NAFLD
A recent trend of NAFLD research is the phenotypic classification of NAFLD. Of the various phenotypes, much attention has been focused on non-obese or lean NAFLD. Non-obese NAFLD literally refers to fatty liver disease that occurs in the absence of overweight or obesity [54]. Clinical, metabolic, and histological phenotypes of non-obese NAFLD are similar to those of typically obese NAFLD since metabolic disease and insulin resistance are frequently accompanied by non-obese NAFLD [54-56]. The prevalence of non-obese NAFLD ranges from 8.7% to 12.4%, and is especially high in Eastern compared with Western countries [57,58]. Many earlier studies from Eastern countries have reported the incidence and characteristics of non-obese NAFLD, and similar findings have been found in Western populations. Based on these results, the concept of ‘metabolically obese normal weight’ (MONW) has been suggested, and novel pathophysiological mechanisms of MONW or non-obese NAFLD are emerging. Non-obese NAFLD is usually defined as a BMI < 30 in the Western population and < 25 in the Eastern population with; cutoff value of BMI for lean NAFLD is 25 in Western population and 23 in Eastern population [54].
Epidemiology
Whether non-obese NAFLD differs from obese NAFLD in terms of phenotypic and etiological aspects of NAFLD, unique pathophysiology, and clinical prognosis remains unclear. Numerous epidemiological studies have continued to report this unique disease entity beyond ethnicity and locality [59]. In Korea, the prevalence of non-obese NAFLD was approximately 12.6% of the total population in 2012, and the worldwide prevalence of non-obese NAFLD varies up to 30% (7%–21% in the West, 3%–27% in the East) [54,60]. BMI does not provide accurate information about the distribution of body fat [61]. When fat tissue dominantly accumulates in the abdominal visceral organ, non-obese NAFLD is likely to occur even in normal weight. It is well known that the waist circumference and the waist circumference to pelvic circumference ratio are more consistently correlated with the visceral fat mass than the BMI itself [62]. In addition, because of racial differences in body fat distribution, a new standard for non-obese NAFLD is needed in Asia, including Korea [63,64].
Pathogenesis
The pathogenesis of non-obese NAFLD has been found to be similar to that of obese NAFLD due to visceral obesity, dietary preference, and insulin resistance [60]. However, other factors seem to exist, including genetic polymorphism or developmental processes. Recently, there have been many studies to classify various phenotypes of NAFLD using genetic risk factors. The best-known gene is PNPLA3 , and other genes such as CETP, SREBF, and TM6SF2 have also been found to be associated with non-obese NAFLD. Recent genome-wide association studies (GWAS) reported genetic polymorphisms of NAFLD and body fat distribution in non-obese NAFLD, and these findings may contribute to identify the cause of non-obese NAFLD when integrated with previous genetic results [65,66].
Palatin-like phospholipase domain containing 3 (PNPLA3)
Hepatic fat accumulation has been reported to be associated with PNPLA3 polymorphism, and PNPLA3 variation confers susceptibility to NASH, fibrosis, and HCC [67-69]. PNPLA3 variant has been reported to increase the prevalence of NAFLD independent of insulin resistance or other comorbidities such as diabetes and dyslipidemia, which are the main features of NAFLD. Many studies are currently in progress, especially for non-obese NAFLD [70,71]. In a study in Hong Kong, PNPLA3 variant was found in 78.4% of the non-obese NAFLD group and in 59.8% of the obese NAFLD group [72]. The racial and ethnic differences of non-obese NAFLD have not been clarified and merit needs further investigation.
Cholesteryl ester transfer protein (CETP)
CETP encoded by the CETP gene is an important protein in transportation of cholesterol from the peripheral tissue to the liver [73]. Among the genetic variants of CETP, two single-nucleotide polymorphisms, rs12447924 and rs12597002, increase the prevalence of NAFLD, especially non-obese NAFLD [74]. In individuals homozygous for the nonvariant, the prevalence of NAFLD is only 3%–5%, whereas homozygous expression of the risk variant tends to increase the prevalence of NAFLD to 25%–33%.
Sterol regulatory element-binding factor (SREBF)
SREBP plays an important role in the synthesis, uptake, and secretion of intracellular cholesterol [75]. Increased expression of SREBF-2 is associated with more severe NAFLD in terms of histological examination [76]. In particular, the expression of SREBF is increased in non-obese and non-diabetic patients, and patients with SREBF expression are more likely to progress to NASH over the long term compared to those without it (odds ratio 2.9; 95% confidence interval 2.1–4.2) [77].
Prognosis and treatment
So far, studies on prognosis confined to non-obese NAFLD are lacking, but the prognosis of non-obese NAFLD does not seem to be different from that of obese NAFLD. Since many predictive models for noninvasive estimation of NASH and advanced fibrosis are validated mainly in obese patients, tailored predictive models targeting non-obese NAFLD are warranted to realize precise and personalized medicine in NAFLD [78-80]. Lifestyle modification, such as regular exercise and diet control to achieve weight loss, is the cornerstone of treatment in non-obese NAFLD as well as obese NAFLD. However, in non-obese NAFLD, remarkable changes are not likely to be achieved through improved lifestyle modifications. It has been reported that the intake of unsaturated fats is significantly lower in non-obese NAFLD patients [81]. In contrast, a recent study from Korea reported that obese NAFLD patients had lower intake of unsaturated fats and higher levels of animal fat, and that exercise levels of <2 hours per week and excessive carbohydrate intake were associated with non-obese NAFLD [82]. Therefore, it is desirable to maintain moderate exercise intensity and reduce carbohydrate intake in non-obese NAFLD patients. Currently, there is no effective pharmacotherapy for non-obese NAFLD patients. Furthermore, since most clinical trials involving drug development for NAFLD or NASH target patients with severe obesity, it is unclear whether the therapeutic approach targeted by these drugs will be equally applicable to non-obese NAFLD patients.
CONCLUSION
For a successful NASH trial, it is advisable to use a validated endpoint. Many surrogate markers that can replace liver biopsy are currently available and widely used, especially in phase II trials. In phase III trials, histological examination is essential and complete remission of NASH without worsening of liver fibrosis is considered the optimal endpoint. There is no formally approved drug for the treatment of NASH but several clinical trials, including large-scale phase III trials, are underway. Recently, research on genetic variants that determine various phenotypes of NAFLD, such as non-obese NAFLD, has been conducted to reveal the pathogenesis of non-obese NAFLD/NASH, and it is expected that individualized treatments for non-obese NAFLD patients will be realized soon.
Notes
Author contributions
J. J. Yoo and W. Kim conceptualized and designed the study, drafted the initial manuscript, and reviewed and revised the manuscript. M.Y. Kim, and D.W. Jun contributed to study design, data collection and review of the manuscript. S.G. Kim was involved in interpretation of data. J.E. Yeon and J.W. Lee revised the article critically for important intellectual content. Y.K. Cho, S.H. Park and J. H. Sohn were responsible for the study conception and design, as well as the intellectual content of the paper. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the word.
Conflict of Interest
The authors declare they have no conflict of interest.
Abbreviations
ALT
alanine aminotransferase
AST
aspartate aminotransferase
BMI
body mass index
CAP
controlled attenuation parameter
ELF
enhanced liver fibrosis
FIB4
fibrosis-4
GWAS
genome-wide association studies
HbA1c
glycosylated hemoglobin
HCC
hepatocellular carcinoma
HOMA-IR
homeostasis model assessment for insulin resistance
HVPG
hepatic venous pressure gradient
MELD
model for end-stage liver disease
MONW
metabolically obese normal weight
MRE
magnetic resonance elastography
MRI-PDFF
magnetic resonance imaging–derived proton density fat fraction
NAFLD
nonalcoholic fatty liver disease
NAS
NAFLD activity score
NASH
nonalcoholic steatohepatitis
NASH CRN
Nonalcoholic Steatohepatitis Clinical Research Network
NFS
NAFLD fibrosis score
TE
transient elastography