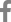INTRODUCTION
Hepatitis C virus (HCV) is an RNA virus that is unable to integrate into the host genome. However, its proteins interact with various host proteins and induce host responses that potentially contribute to the malignant transformation of cells. Hepatocellular carcinoma (HCC) development is usually a final consequence of sequential progression of chronic fibrosing liver diseases, and HCC usually occurs only after establishment of liver cirrhosis in HCV-infected individuals.1 In cirrhotic patients with HCV infection, the annual HCC development rates range between 1-7%.2
The incidence of HCV-related HCC continues to rise and is estimated to remain high in the next two decades.3 Although epidemiological evidence has suggested a clear, close relationship between HCV infection and HCC,4,5 the prevalence of HCV infection in HCC patients differs noticeably between geographical regions. HCV infection is found in 70-80% of HCC patients in Japan, 70% in Egypt, 40-50% in Italy and Spain, about 20% in the United States and Korea, and less than 10% in China.6-8 HCV increases the risk of HCC by promoting inflammation and fibrosis of the infected liver that eventually results in liver cirrhosis. Other factors including alcohol intake, diabetes, and obesity have also been reported to increase the risk of HCC development by about two- to fourfold, indicating a strong life-style effect on the process of hepatocarcinogenesis.9,10
Recent genome-wide association studies (GWAS) have suggested that the natural course of HCV infection might be modified by the genetic background of the host.11,12 Thus, both host and virus factors are considered to affect the process of hepatocarcinogenesis in a complex manner.
In this review, we summarize the current knowledge of the mechanisms of hepatocarcinogenesis induced by HCV infection.
Molecular pathways in hepatocarcinogenesis
HCC is a highly heterogeneous tumor. Hepatocarcinogenesis is a complex multistep process involving a number of genetic and epigenetic alterations, the activation of cellular oncogenes and/or the inactivation of tumor suppressor genes, and dysregulation of multiple signal transduction pathways. These pathways include Wnt/╬▓-catenin, p53, pRb, Ras, mitogen-activated protein kinase (MAPK), Janus kinase (JAK)/signal transducer and activator of transcription (STAT), phosphatidylinositol 3-kinase (PI3K)/Akt, Hedgehog and growth factors such as epidermal growth factor, and transforming growth factor-╬▓ (TGF-╬▓) pathways.13-15
Fibrosis and hepatocarcinogenesis
The vast majority (80-90%) of HCCs develop in a cirrhotic liver.16 During the progression of liver injury, hepatic stellate cells (HSCs) become activated, losing retinoid-containing lipid droplets and transforming into myofibroblast-like cells, which produce extracellular matrix, the first step in hepatic fibrosis.17 Unchecked progression of fibrosis ultimately eventuates in irreversible cirrhosis. The activated HSCs become responsive to both proliferative platelet-derived growth factor (PDGF)18 and fibrogenic (TGF-╬▓) cytokines,19 which are upregulated in fibrogenesis and modulate inflammatory signaling from infiltrating immune cells.20 PDGF can activate both MAPK and PI3K/Akt signaling cascades.20 In PDGF-C transgenic mice, activation and proliferation of HSCs precedes development of fibrosis, which in turn is followed by the occurrence of HCC. This progression is analogous to that seen in human HCC.21 The cirrhotic liver is also associated with telomere shortening, which may in turn lead to chromosomal instability and deletion of check points.22 Increased survival factors that prevented apoptosis of DNA-damaged hepatocytes and activated stellate cells (for example, Gas6215) and reduced tumor surveillance function due to decreased natural killer cell function are all possible factors related to HCC development in cirrhosis.23 Recent studies have found that stellate cells express stem cell markers such as CD133, nestin, c-kit and p75 neurotrophin receptor,24-27 and activated stellate cells appear to contribute to the stem cell niche.28 Hedgehog and Wnt signaling pathways involved in stem cell differentiation and cancer formation are also found in stellate cells.29,30 These lines of evidences suggest that stellate cells may harbor the potential to transdifferentiate into progenitor cells and possibly be linked to the development of HCC.23
Virus proteins and host responses
HCV belongs to the Flaviviridae family. It has a 9.6-kb positive-stranded linear RNA genome containing 5' and 3' untranslated regions including control elements required for translation and replication. The untranslated regions flank an uninterrupted open-reading frame encoding a single polyprotein of 3010 or 3011 amino acids, which is processed into three structural (core, E1, and E2) and seven non-structural (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins by host and viral proteases.31 HCV is an RNA virus unable to reverse transcribe its genome and thus to integrate it into the host genome. Instead, viral proteins and their evoked host responses contribute mostly to the viral oncogenic processes.
Core protein
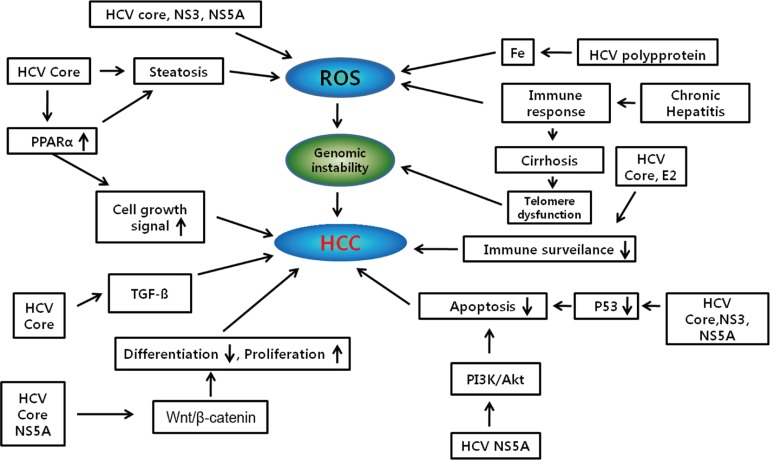
HCV core protein has been proposed to be involved in apoptosis, signal transduction, reactive oxygen species (ROS) formation, lipid metabolism, transcriptional activation, transformation and immune modulation (Fig. 1).15,32
Several recent studies have indicated the statistically significant high frequency of mutations in the core gene in HCV-infected patients who developed HCC.33,34
HCV core protein binds to several tumor suppressor proteins, including p53, p73 and pRb.35,36 HCV core interacts with p73, causes nuclear translocation of core protein and prevents p73 ╬▒-dependent cell growth arrest in a p53-dependent manner.37 HCV core can also modulate the expression of the cyclin dependent inhibitor p21WAF1, which is a major target of p53 and regulates the activities of cyclin/cyclin-dependent kinase complexes involved in cell-cycle control and tumor formation.38,39 Core protein may also influence the growth and proliferation of host cells through activation of signaling pathways such as Raf/MAPK,40 Wnt/╬▓-catenin,41 and TGF-╬▓.15,42 These pathways are known to be activated in HCC.43 However, the functional relevance of mutant core proteins on the malignant transformation of hepatocytes or the HCV life cycle has yet to be clarified.
NS3 protein
NS3 inhibits the activity of the p21WAF1 promoter in a dose-dependent manner and is synergistic with core in this regard.46 NS3 inhibits the function of p53 in an NS3 sequence in an NS3 sequence-dependent manner.47 The expression of NS3 enhances cell growth, JNK activation and DNA-binding activities of the transcription factors AP-1 and ATF-2.48 NS3 also induces TNF-a production by activation of AP-1 and NF-kB.49
NS5A protein
NS5A is essential for the replication of the HCV genome and is localized mainly in the cytoplasm of infected cells in association with the endoplasmic reticulum (ER). NS5A is involved in a large number of cellular functions, including apoptosis, signal transduction, transcription, transformation and ROS production. High frequencies of wild-type NS5A genes were reported to be dominant in liver cirrhosis patients who finally developed HCC compared with those who did not,50 but the mechanistic significance of the NS5A wild/mutant genotypes in the process of HCV-related hepatocarcinogenesis remains uncertain. NS5A protein has been suggested to interact with various signaling pathways including cell cycle/apoptosis51 and lipid metabolism52-54 in host cells and shares some signaling targets with core protein. NS5A is recognized as a transcriptional activator for many target genes55 including p53 and its binding protein, TATA binding protein (TBP). Transcription factor IID activities were reported to be modified by NS5A in the suppression of p53-dependent transcriptional transactivation and apoptosis.56,57 NS5A may also interact with pathways such as Bcl2,58 PI3-K,59 Wnt/╬▓-catenin signaling,60 and mTOR61 to activate cell proliferation signaling and inhibit apoptosis. Taken together, intriguing data concerning the function of core and NS5A proteins on host cell signaling pathways, transcriptional activation, apoptosis, oxidative stress, and lipid metabolism suggest a diverse role for HCV proteins in the pathophysiology of chronic HCV infection that leads to malignant transformation in infected hepatocytes.
Genomic characteristics of HCV related HCC
It is now widely believed that tumors originate from normal cells as a result of accumulated genetic/epigenetic changes. These alterations affect the signaling pathways at transcriptional and posttranscriptional level that drive cells into uncontrolled cell division, growth, and migration. HCV proteins in infected cells can cause various host responses at transcriptional/translational/posttranslatonal levels, so genetic/genomic alterations and transcriptional/translational modifications can ultimately affect the cellular signaling pathway at the transcriptional level.
Recent advancement of molecular technologies have yielded comprehensive gene expression profiling techniques that have successfully provided candidate diagnostic and prognostic markers in human cancers.
Over the past decade, several methods (including differential display, serial analysis of gene expression [SAGE], and microarray) have been developed to allow comparative studies of gene expression between normal and cancer cells on a genome-wide scale,62 and the analysis of a set of all RNA molecules (mainly indicating messenger RNAs [mRNAs]) is termed as whole transcriptome analysis.
Early microarray and SAGE studies investigating the gene expression patterns of chronic hepatitis B (CHB) and CHC indicated that many genes were differentially regulated between hepatitis B and C. In CHB, genes for induction of apoptosis, cell cycle arrest, and extracellular matrix degrading were up-regulated, whereas in hepatitis C, genes with antiapoptotic effects, cell cycle acceleration, and extracellular matrix storage were up-regulated.63,64
An early study comparing genes activated in HCV-related and HBV-related HCCs showed that expression of genes encoding CYP2E, AKR1C4, EPHX1, and FMO3 enzymes that convert several pro-carcinogens to activated metabolites increased exclusively in HCV-positive HCCs, which may suggest that their enhanced expression leads to a greater contribution of carcinogenic metabolites to the mechanisms of HCV-specific hepatocarcinogenesis. On the other hand, decreased expression of detoxification enzymes including UGT1A1, UGT2B10, and GPX2 was noted in HBV-positive HCCs. These results suggest that decreased expression of detoxification enzymes may be involved especially in the mechanisms of HBV-specific hepatocarcinogenesis.
The genes associated with xenobiotic metabolism were more abundantly expressed in HCV-related HCC, suggesting a detoxification role, which is potentially induced by chronic inflammation and generation of ROS resulting from HCV infection.65 In contrast, HBV-related HCC might closely correlate with the activation of imprint genes, including insulin-like growth factor-II (IGF-II), suggesting a role of de-differentiation or epigenetic alteration of the host genome in HBV-related HCC. The expression levels of many detoxification-related genes were increased in HCV-related HCC in comparison to HBV-related HCC. Markedly reduced levels of detoxification-related genes in HBV-related HCC suggests that HBV-infected liver could be more susceptible than HCV-infected liver to various xenobiotics or carcinogens.66 Activation of genes associated with interferon, oxidative stress, apoptosis, and lipid metabolism signaling was detected in HCV-related HCC and CHC specimens,64,67,68 consistent with numerous functional studies that have investigated the host response evoked by HCV structural and non-structural proteins.51
HCC risk predictors that identify the subset of cirrhotic patients with the highest risk of HCC are sorely needed. In addition, identification of molecular biomarkers may open new prospect toward the discovery of therapeutic targets.
Transcriptome analysis has also recently gave new understanding on the transcriptional alteration events occurring in early stages of HCV-related hepatocarcinogenesis. GPC3 (encoding Glypican 3) was suggested as one of the most activated transcripts in the early stage of hepatocarcinogenesis,64,69 also several recent studies have reported that gene signatures including GPC3 can successfully discriminate HCCs from pre-malignant dysplastic nodules and cirrhosis nodules.70,71
The genetic approach between each of the stages from normal, cirrhotic, and dysplastic to early and advanced HCV-related HCC identified gene signatures that accurately reflect the pathological progression of disease at each stage. In addition, pathway analysis revealed dysregulation of the Notch and Toll-like receptor pathways in cirrhosis, followed by deregulation of several components of the JAK/STAT pathway in early carcinogenesis, then upregulation of genes involved in DNA replication and repair and cell cycle in late cancerous stages.50,72 These findings provide a comprehensive molecular portrait of genomic changes in progressive HCV-related HCC. Aimed at identifying etiology-specific or independent genetic variants predictive of HCC risk, efforts have focused on the search for single nucleotide polymorphisms (SNPs) associated with the presence of HCC in candidate genes such as epidermal growth factor (EGF) based on certain biological hypothesis.73
Recent development of high-throughput genomics technology has enabled genome-wide scans of such loci in the setting of a GWAS. In HCC, the first GWAS was conducted on hepatitis B-related HCC patients and identified a SNP possibly associated with altered expression and function of several potential tumor suppressor genes in 1p36.22 namely KIF1B, UBE4B, and PGD.74
The first GWAS on HCV-related HCC has recently been reported by Kumar and colleagues.75 By analyzing 721 patients with HCV-related HCC and 2890 HCV-negative controls of Japanese origin for 432,703 autosomal SNPs, they identified eight possible HCC susceptibility loci with modest statistical significance. The following replication stage, involving 673 independent cases and 2596 HCV-negative controls, confirmed a novel SNP rs2596542 located in the 50 flanking region of MICA, the MHC class I polypeptide-related sequence A gene, on chromosome 6p21.33.
The authors further genotyped the locus in additional 1730 individuals with CHC who had not developed liver cirrhosis, and found that the association of the risk allele was observed in the comparison between CHC and HCC patients, but not in the comparison between CHC patients and HCV-negative controls, suggesting that the SNP is associated with progression from CHC to HCC rather than susceptibility to HCV infection.
However, the study by Kumar et al75 did not use HCV-related cirrhosis without HCC as the controls, it is possible that the risk allele in the MICA gene is actually responsible for increased progression of liver cirrhosis, which eventually contributes to development of HCC. That is, information from this allele may not be useful in distinguishing HCC high risk population among HCV related cirrhotic patients. In fact, the soluble MICA protein level was not different between CHC, cirrhosis, and HCC patients. This needs to be clarified in future studies for example by genotyping patients with HCV-related cirrhosis and following for HCC development to evaluate the risk allele's association with hazard of HCC occurrence within cirrhotic patients.76
Authors analyzed a large set of HCV Japanese carriers (n=3,312) using a case-control design, and they interrogated 467,538 germline SNPs and identified one, rs1012068, significantly associated with the risk of developing HCC. The SNP is located in chromosome 22, and by using fine mapping studies the authors identified DEPDC5 as the target gene harboring the different genotypes. Despite the function of this gene is unknown, there is some evidence of aberrations affecting its locus in human cancer (e.g., glioblastoma).
Recent advances in transcriptome analysis have also provided detailed information on the status of small noncoding RNAs, microRNAs (miRNAs) that regulate gene expression by targeting mRNAs through translational repression or RNA degradation. Many fundamental biological processes are modulated by miRNAs, and an important role for miRNAs in carcinogenesis is emerging.
Although the mechanisms of altered miRNA levels in human cancer are quite varied, including deletions, amplification or mutations involving miRNA genes, it is clear that miRNA-regulated expression of oncogenes and tumor suppressor genes contribute to most - if not all - human cancers.1 Earlier studies noted specific changes in miRNA expression patterns in HCC as compared with adjacent normal liver tumor tissues, or liver cirrhosis that correlated with the disease outcome (Table 2).3,81-84
Since there is no HCV encoded oncoprotein, the question arises whether the deregulated miRNAs in HCC serve as "oncomiRs," that could function as an oncogene or a tumor suppressor, to regulate cell proliferation by targeting cell cycle check points and/or growth factors. Such oncomiRs in liver cancer would be expected to be involved through each step from normal liver to cirrhosis to HCC. The extant literature strongly supports the role of specific oncoMirs in the development and maintenance of HCC.
More recently, the investigators focused on miR-26a whose expression is most significantly perturbed in MYC-induced liver cancer model.87 MiR-26a targets expression of cyclins D2 and E2; and ectopic expression of miR-26a induced G1 arrest in HepG2 HCC cell line. Examination of paired biopsies from normal human liver tissues as compared with liver cancer showed consistent reduction of miR-26a in liver cancer, while miR-26a is expressed at high levels in normal liver as well as other tissues. Since miR-26a induces G1 arrest by targeting cyclins D2 and E2, the authors reasoned that forced expression of miR-26a in liver cancer cells might arrest tumor growth.
Indeed, the systemic administration of miR-26a in a mouse model of HCC using adeno-associated virus vector system, resulted in the inhibition of cancer cell proliferation by inducing tumor-specific apoptosis, with dramatic protection from disease progression without toxicity.87
Thus, the delivery of miR-26a, which is highly expressed and therefore tolerated in normal, but not in liver cancer cells, may be a useful strategy for miRNA-replacement therapy for HCC.89
Expression of miRNAs including miR-122 and -199a has been reported to modulate HCV replication,3,85,90 and miR-122 expression can be regulated by host interferon signaling and responses.52 HCV induced miR-155 expression promotes hepatocyte proliferation and tumorigenesis by activating Wnt signaling. The overexpression of miR-155 significantly inhibited hepatocyte apoptosis and promoted cell proliferation.91
HCV protein expression in turn could induce miRNAs and might affect the tumor suppressor DLC1 and the chemosensitivity of malignantly transformed cells.53,88 Several miRNAs were also differentially expressed between HCV-related and HBV-related HCCs as well as their corresponding non-cancerous liver tissues. The candidate signaling pathways potentially altered by miRNAs in HCV-related tissues were those associated with antigen presentation, cell cycle, and lipid metabolism,92 consistent with the mRNA microarray data described above. MiRNAs have also recently been reported to successfully discriminate between HCC and cirrhotic liver tissues,55 implicating their role in the early stages of malignant transformation. These data suggest that miRNAs may be good targets for the eradication of HCC as well as hepatocytes infected with HCV.
CONCLUSION
Hepatocarcinogenesis is a multistep process and involves multiple cellular signaling pathways. Although HCV is the major risk factors leading to the development of HCC, the precise pathogenetic mechanisms linking viral infection and HCC remain uncertain. Viral proteins also have been implicated in disrupting several cellular signal transduction pathways that affect cell survival, proliferation, migration and transformation (Fig. 2).15 Current advances in gene expression profile and selective mRNA analysis have improved approach to the pathogenesis of HCC. The heterogeneity of genetic events observed in HCV-related HCCs has suggested that complex mechanisms underlie malignant transformation induced by HCV infection. Considering the complexity and heterogeneity of HCCs of both etiological and genetic aspects, further molecular classification is required and an understanding of these molecular complexities may provide the opportunity for effective chemoprevention and personalized therapy for HCV-related HCC patients in the future.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print