Hepatitis C virus: virology and life cycle
Article information
Abstract
Hepatitis C virus (HCV) is a positive sense, single-stranded RNA virus in the Flaviviridae family. It causes acute hepatitis with a high propensity for chronic infection. Chronic HCV infection can progress to severe liver disease including cirrhosis and hepatocellular carcinoma. In the last decade, our basic understanding of HCV virology and life cycle has advanced greatly with the development of HCV cell culture and replication systems. Our ability to treat HCV infection has also been improved with the combined use of interferon, ribavirin and small molecule inhibitors of the virally encoded NS3/4A protease, although better therapeutic options are needed with greater antiviral efficacy and less toxicity. In this article, we review various aspects of HCV life cycle including viral attachment, entry, fusion, viral RNA translation, posttranslational processing, HCV replication, viral assembly and release. Each of these steps provides potential targets for novel antiviral therapeutics to cure HCV infection and prevent the adverse consequences of progressive liver disease.
INTRODUCTION
Hepatitis C virus (HCV) is a hepatotropic RNA virus of the genus Hepacivirus in the Flaviviridae family, originally cloned in 1989 as the causative agent of non-A, non-B hepatitis.1,2 It causes acute and chronic hepatitis in humans and chimpanzees with a high propensity for chronicity. If untreated, chronic hepatitis C can progress to cirrhosis and hepatocellular carcinoma in a subset of patients.3 Until recently, the standard of care for patients with chronic hepatitis C involved dual therapy with pegylated interferon (IFN) alpha and ribavirin (PEG IFN/riba) in most countries. Dual PEG IFN/riba therapy achieved sustained virological response (SVR) in only 50% of patients infected with the more common HCV genotype (genotype 1) compared to 80% SVR rate in patients infected with HCV genotype 2 or 3.4 Moreover, combined PEG IFN/riba therapy is costly and prolonged (e.g. 24-48 weeks) with numerous adverse effects that are difficult to tolerate. In 2011, two inhibitors of the virally encoded NS3/4A protease became available as a part of standard therapy in some countries, especially against HCV genotype 1. Triple therapy combining one of these first-generation protease inhibitors with PEG IFN/riba therapy has improved SVR rate from around 50% to 70% in some clinical trial cohorts,5-8 However, this new regimen has limited efficacy in certain special populations (e.g. cirrhotic patients, transplant recipients, primary non-responders and hemodialysis patients) due to underlying IFN resistance, emergence of protease inhibitor resistance mutations and/or increased drug toxicity. Thus, there are ongoing efforts towards better therapeutic options with shorter treatment duration with less toxicity and drug resistance-preferably as IFN-free, all oral combination regimens.
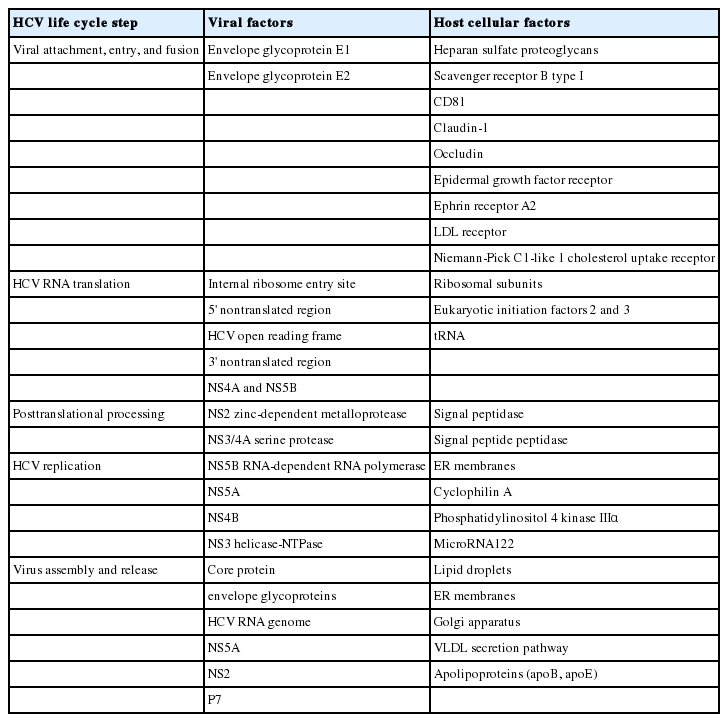
The recent HCV therapeutic development has been greatly enhanced by basic understanding of HCV virology and life cycle, through studies using HCV cell culture systems and replication assays. In this article, we review various steps in HCV life cycle that also serve as relevant targets for potential novel therapeutics, including viral attachment, entry, fusion, viral RNA translation, posttranslational processing, HCV replication, viral assembly and release (Fig. 1).9

HCV genome and its products
HCV is a positive-sense, single-stranded enveloped RNA virus approximately 9600 nucleotides in length. Approximately 1012 viral particles are generated daily in chronically HCV-infected patient.10 Due to the highly error prone RNA polymerase, HCV also displays remarkable genetic diversity and propensity for selection of immune evasion or drug resistance mutations.11 There are 6 major HCV genotypes (numbered 1-6) that vary by over 30% in nucleotide sequence from one another.12 The HCV genome has one continuous open reading frame flanked by nontranslated regions (NTRs) at 5' and 3' ends. The HCV 5'NTR contains 341 nucleotides located upstream of the coding region and is composed of 4 domains (numbered I to IV) with highly structured RNA elements including numerous stem loops and a pseudoknot.13,14 The 5' NTR also contains the internal ribosome entry site (IRES), that initiates the cap-independent translation of HCV genome into a single polyprotein15 by recruiting both viral proteins and cellular proteins such as eukaryotic initiation factors (eIF) 2 and 3.16-18
The HCV open reading frame contains 9024 to 9111 nucleotides depending on the genotype. It encodes a single polyprotein that is cleaved by host and viral proteases into 10 individual viral proteins with various characteristics.19
Structural proteins
HCV core is the viral nucleocapsid protein with numerous functionalities involving RNA binding, immune modulation, cell signaling, oncogenic potential and autophagy. HCV core protein also associates with the lipid droplets where HCV assembly also takes place. HCV E1/E2 are glycosylated envelope glycoproteins that surround the viral particles. HCV envelope is targeted by virus neutralizing antibody selection pressure with high degree of sequence variation that may render antibody responses ineffective and contributes to HCV persistence.20-22 The small ion channel protein p7 is downstream of the envelope region and is required for viral assembly and release.
Nonstructural proteins
NS2 is the viral autoprotease that plays a key role in viral assembly, mediating the cleavage between NS2 and NS3. NS3 encodes the N-terminal HCV serine protease and C-terminal RNA helicase-NTPase. NS3 protease play a critical role in HCV processing by cleaving downstream of NS3 at 4 sites (between NS3/4A, NS4A/4B, NS4B/NS5A, NS5A/NS5B). It also cleaves the TLR3 adaptor protein TRIF and mitochondrial antiviral signaling protein MAVS, thereby blocking the cellular type I IFN induction pathway.23 NS3 is one of the key targets for HCV antiviral drug development. NS4A forms a stable complex with NS3 and is a cofactor for NS3 protease. The role of NS4B is not well understood, although it is known to induce the membranous web formation. NS5A is a dimeric zinc-binding metalloprotein which binds the viral RNA and various host factors in close proximity to HCV core and lipid droplets. Inhibitors of HCV NS5A showed antiviral effect in patients and are in rapid clinical development. Finally, NS5B is the RNA-dependent RNA polymerase (RdRp) which is also being actively targeted for antiviral drug development.
Collectively, these proteins also contribute to various aspects of HCV life cycle, including viral attachment, entry and fusion, HCV RNA translation, posttranslational processing, HCV replication, virus assembly and release.
HCV virion and lipoviroparticle
The HCV viral particle includes HCV RNA genome, core and the envelope glycoproteins, E1 and E2.2 HCV RNA genome interacts with the core protein to form the viral nucleocapsid, in association with cytosolic lipid droplets (cLD). The viral nucleocapsid is enveloped in lipid-rich viral envelope with the E1 and E2 glycoproteins that play key roles in virus entry through receptor binding and fusion.24,25 E1/E2 glycoproteins are type I transmembrane glycoproteins that can form non-covalent heterodimers within the infected cells or large covalently linked complexes on the viral particle. They include a large N-terminal ectodomain and a short C-terminal transmembrane domain. The transmembrane domains are involved in membrane anchoring, endoplasmic reticulum (ER) localization, and heterodimerization of the envelope glycoproteins.26-28 E1 and E2 are highly glycosylated, containing up to 6 and 11 glycosylation sites, respectively.29
Hypervariable regions have been identified in the E2 envelope glycoprotein.30 In particular, hypervariable region 1 (HVR1) is a 27 amino acids long segment of basic residues with a high degree of sequence variability as well as highly conserved conformation, suggesting a role as HCV neutralizing epitope and involvement with host entry factor.31-33 HCV virion also associates with various lipoproteins such as apoE, apoB, apoC1, apoC2 and apoC3 to form a complex lipoviroparticle (LVP) and various lipoprotein components can influence HCV entry.34
Viral attachment, entry and fusion
Viral entry into the host cell involves a complex series of interactions including attachment, entry and fusion. The initial viral attachment to its receptor/co-receptors may involve HVR1 in HCV E235,36 with facilitation by heparan sulfate proteoglycans expressed on hepatocyte surface.37-40 While LDL receptors (LDLR) can bind HCV and promote its cellular entry,41 HCV-LDLR interaction may be non-productive and can potentially lead to viral particle degradation.40 Following attachment to the entry factors, HCV is internalized into the target cells via a pH-dependent and clathrin-mediated endocytosis.42-45
Multiple cellular receptors and entry factors for HCV have been identified, including the scavenger receptor class B type I (SRB1),46 and CD8147 as well as tight junction proteins, claudin-1 (CLDN1)48 and occludin (OCLN).49,50 Additional recently identified entry factors include the receptor tyrosine kinases (RTK) epidermal growth factor receptor (EGFR), ephrin receptor A2 (EphA2)51 and Niemann-Pick C1-like 1 cholesterol absorption receptor (NPC1L1).52 The various entry factors are briefly described below:
Scavenger receptor class B type I
The first two HCV entry factors (SRB1 and CD81) were identified as binding partners of HCV E2.46,47 SRB1 is a 509-amino acid multi-ligand glycoprotein receptor which is known to be a major receptor for high-density lipoproteins (HDLs).53 It is highly expressed in the liver and promotes selective uptake of HDL cholesterol esters into hepatocytes.54 A role of SRB1 in HCV entry was first suggested by its binding of HCV E2 through HVR1.46 The lipid transfer activity of SRB1 may be required for HCV cell entry since lipoprotein ligands can modulate HCV/SRB1 interactions with HCV entry enhancement by HDL and inhibition by oxidized LDL.54-57 Of interest, level of SRB1 on hepatocytes can regulate the level of HCV entry and infectivity,58 whereas steroids such as prednisolone can promote HCV entry by up-regulating SRB1 expression.59 Human monoclonal antibody against SRB1 was shown to prevent HCV infection in vitro in hepatocytes and in vivo using chimeric mice engrafted with human hepatocytes.57 In a phase I clinical trial of HCV-infected patients, SRB1 antagonist was shown to inhibit HCV replication with added synergy for other antiviral therapeutics.60
CD81
Human CD81 is a tetraspanin molecule that is broadly expressed and involved in many cellular functions including adhesion, morphology, proliferation and differentiation. It includes 4 transmembrane domains, 2 short intracellular domains and two extracellular loop domains (small and large).54 CD81 is likely involved after the very early phase of HCV entry (i.e. after SRB1), promoting a conformational change in the HCV E1/E2 envelope glycoprotein to facilitate low pH-dependent fusion and viral endocytosis.61 Involvement at a later phase after virus internalization was also suggested recently.62 CD81 large extracellular loop binds HCV through its envelope glycoprotein E2.47,63 The sequence of CD81 large extracellular loop is conserved between humans and chimpanzees, the only 2 species permissive to HCV infection in vivo.64,65 However, CD81 from other species (e.g. African green monkey, tamarin, rats, mice, hamster) can support entry of HCV clones in vitro, suggesting that CD81 polymorphism is not sufficient to define HCV susceptibility.66 Although the ubiquitous expression of CD81 in multiple cell types may limit focused therapeutic application, anti-CD81 was shown to inhibit HCV entry in vivo in chimeric mouse model of HCV replication.67
The tight junction proteins Claudin 1 and Occludin
Two tight junction proteins (CLDN1 and OCLN) identified by screening cDNA library for cellular factors that enable infection by HCV pseudoparticles were shown to be essential entry factors for HCV.48,50 Interestingly, neither CLDN1 nor OCLN seems to interact directly with HCV particles. However, CLDN1 may interact with CD81 as a part of the HCV receptor complex,68,69 it is believed that CLDN1 and OCLN are involved in a later phase of HCV entry, after SRB1 and CD81, although their precise roles are not fully defined. It was also shown that HCV envelope glycoproteins promote coendocytosis of CD81 and CLDN1 in a clathrin- and dynamin-dependent process.62 Finally, anti-CLDN1 monoclonal antibodies were shown to inhibit HCV infection in hepatocytes in vitro by neutralizing the interactions between HCV E2 and CLDN1.70,71
Receptor tyrosine kinases and Niemann-Pick C1-like 1 cholesterol absorption receptor
Two RTKs including the EGFR and EphA2 were recently identified as HCV entry factors by a functional RNAi kinase screen.51 These RTKs appear to participate in HCV entry after the initial attachment/binding step, by regulating CD81 and CLDN1 co-receptor interactions and membrane fusion. RTKs also contributed to cell-cell transmission of HCV. Relevant for clinical application, infection by all major HCV genotypes and escape variants could be inhibited by RTK-specific ligands and antibodies including erlotinib (an EGFR-specific antibody that is FDA-approved for cancer therapy). Finally, a group in Chicago identified NPC1L1 (a cholesterol sensing receptor expressed on apical surface of hepatocytes as well as enterocytes) as a new HCV entry factor, which can be inhibited by an available NPC1L1 antagonist ezetimibe which is FDA-approved to treat hypercholesterolemia.52
Collectively, these receptors and entry factors provide potential avenues to prevent HCV infection and spread, provided that modulation of their physiological role does not lead to significant toxicity.
HCV RNA translation, polyprotein processing and replication
Following target cell entry through receptor-mediated endocytosis, HCV particle undergoes pH-dependent membrane fusion within an acidic endosomal compartment to release its RNA genome into the cytoplasm.19,44,45 HCV polyprotein is translated in rough ER with the positive strand HCV RNA as the template, with the translation initiated in a cap-independent manner via the IRES in the 5'NTR. HCV translation yields a single polyprotein precursor of approximately 3000 amino acid in length, that is further processed by cellular (e.g. signal peptidases) and viral proteases (NS2, NS3/4A) to generate 10 individual viral proteins, including core and envelope glycoproteins, E1 and E2, p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B as mentioned above.
In the course of polyprotein processing, the HCV proteins are seen to be associated with a "membranous web" which includes double-membrane vesicles containing HCV nonstructural proteins, HCV RNA, ER membranes and lipid droplets.19,72,73 The membranous web in HCV-expressing cells appears to be induced by HCV NS4B possibly in combination with NS5A.74,75 Viral RNA replication is believed to occur in these webs with the positive strand RNA genome as a template for the NS5B RdRp to generate the negative strand replicative intermediate, to produce further positive sense genomes. Nascent positive strand RNA genomes can be further translated to produce new viral proteins, or serve as templates for further RNA replication, or be assembled to infectious virions.
Various cellular factors are involved in HCV replication, including cyclophilin A and phosphatidylinositol 4 kinase IIIα (PI4KIIIα). Cyclophilin A can modulate RNA-binding capacity of NS5B polymerase and interact with NS5A. As such, cyclophilin inhibitors have antiviral effect against HCV with clinical development ongoing.76-80 As for PI4KIIIα, it is a lipid kinase that is recruited to the membranous web by NS5A, required for HCV replication and provide integrity to the membranous viral replication complex.81-84
Viral assembly and release
The HCV assembly and release process is not fully understood. However, it appears to be closely linked to lipid metabolism.85 The link between HCV and lipid metabolism was first noted in clinical practice based on fatty changes in liver tissue that was further shown to be associated with HCV core.86 HCV infection induces a profound change in the intracellular distribution of lipid droplets (LDs)87 from generalized cytoplasmic pattern in uninfected cells to accumulation around the perinuclear region in HCV-infected cells associated with viral proteins and genome.88,89 HCV core association with LDs appears to be critical in viral assembly and interventions which block this interaction disrupt virus production.90-92 It is likely that all other viral proteins play a key role in the assembly process, centering around the lipid droplets where assembly is triggered in the membranous and lipid-rich environment by the structural HCV proteins (core/E1/E2/p7/NS2) and the replication complex. The very low density lipoprotein (VLDL) secretion pathway is closely related to that of assembled virions.93,94 The virion is a lipoviroparticle with a lipid composition that resembles VLDL and LDL with associated apoE and/or apoB, which are essential for the infectious virus assembly.93-96 Of clinical relevance, histological fatty changes in the liver has been associated with clinical and therapeutic outcomes97 and HCV genotype 3 was shown to mediate greater steatosis,98 although its pathogenetic mechanisms related to HCV life cycle is not clear. It is also notable that HCV replication can be controlled through cholesterol biosynthetic pathway and lipid-lowering drugs can suppress HCV replication in vitro.99,100
CONCLUSION
Since its initial cloning of HCV, we have gained considerable knowledge about HCV life cycle including entry, translation, replication and host cellular interactions (Table 1).101 These advances have also been critical in antiviral drug development for patients with chronic hepatitis C. After many years, we are now close to being able to cure our patients with potent, pan-genotypic, cost-effective and less toxic therapeutic regimen to prevent their progression to cirrhosis and liver cancer. Remaining challenges include the development of prophylactic vaccine for world-wide application (including countries without sufficient economic development or access to newly developed antivirals) and better understanding of the mechanisms of persistence and pathogenesis of this fascinating virus.
Notes
The authors have no conflicts to disclose.
Abbreviations
cLD
cytosolic lipid droplets
CLDN1
claudin-1
EGFR
epidermal growth factor receptor
eIF
eukaryotic initiation factor
ER
endoplasmic reticulum
EphA2
ephrin receptor A2
HCV
hepatitis C virus
HDLs
high-density lipoproteins
HVR1
hypervariable region 1
IFN
interferon
IRES
internal ribosome entry site
LDs
lipid droplets
LDLR
low density lipoprotein receptor
LVP
lipoviroparticle
NTR
nontranslated region
NPC1L1
Niemann-Pick C1-like 1 cholesterol absorption receptor
OCLN
occludin
PEG IFN
pegylated interferon alpha
PI4KIIIα
phosphatidylinositol 4 kinase IIIα
RdRp
RNA-dependent RNA polymerase
riba
ribavirin
RTK
receptor tyrosine kinases
SRB1
scavenger receptor class B type I
SVR
sustained virological response
VLDL
very low density lipoprotein