INTRODUCTION
Chronic hepatitis B virus (HBV) infection is a major cause of liver inflammation, leading to cirrhosis and hepatocellular carcinoma. Loss of hepatitis B surface antigen (HBsAg) and rise of anti-HBs antibodies in serum have been considered as markers of clearance of HBV replication. With the advent of sensitive detection methods such as polymerase chain reaction (PCR), HBV DNA was detected in about 30-35% of serum samples and 40-50% of liver samples from HBsAg-negative chronic hepatitis and persistence of HBsAg-negative infection is related with liver pathogenesis.1,2,3,4,5,6,7,8 Transmission of HBV infection from such "occult" carriers has also been documented, although infrequently.4
Many studies have attempted to determine the molecular features of HBV persistence in HBsAg-negative patients. Low level viral DNA in the serum and liver is the hallmark of occult HBV infection.9 Sequencing analysis of HBV DNA isolated from HBsAg-negative carriers revealed amino acid substitutions in the major hydrophilic region (MHR) of the viral surface protein, in particular the major B-cell epitope called 'a'-determinant (amino acid residues 124 to 147).10,11,12,13,14 Such viral strains may also contain amino acid changes or insertions in other parts of the surface protein, or substitutions in the PreS domain.11,12,14,15 Coinfection with hepatitis C virus or human immunodeficiency virus has been observed in some cases of occult HBV infection, which may interfere with HBV replication and gene expression thus causing HBsAg negativity.16,17,18
Little is known about the influence of viral strains (HBsAg subtypes or viral genotypes) on spontaneous HBsAg loss. Here we determined the nucleotide sequence encoding the MHR of HBsAg from 25 patients who lost HBsAg during follow up, established impact of some of the MHR mutations on HBsAg expression / secretion by transient transfection experiments, and quantified intrahepatic levels of covalently closed circular (ccc) DNA, the template for viral replication.19 Most patients studied contained low ccc DNA copy numbers. Several mutations detected in our patients reduced HBsAg antigenicity or secretion. Most importantly, majority (75%) of patients who lost HBsAg were infected with genotype D (ayw subtype), a rare genotype in Korea. Only 25% of patients were infected with genotype C (adr subtype), the dominant HBV genotype in this country.
MATERIALS AND METHODS
Patients and samples
A total of 435 chronic HBsAg carriers admitted to the Severance hospital, Yonsei University Medical Center in Seoul, Korea, were followed. All were HBeAg negative for at least a year and had near normal liver function. Those positive for hepatitis C virus infection, with alcohol or drug induced liver diseases, or serum alanine aminotransferase (ALT) levels higher than 1.5 times the upper limit of normal value, were excluded. None of the patients in the "spontaneous loss" group had ever received specific antiviral therapy either before or after HBsAg loss. Serological markers for HBsAg, HBeAg, anti-HBs and anti-HBe were determined by enzyme-linked immunoassays (Behringwerke, Marburg, Germany). Serum HBV DNA was detected by molecular hybridization method using DML 2000 Luminometer (Digene Diagnostics, Silver Spring, USA). The detection limit of this assay is 0.5 pg/mL. All serum samples were reviewed and approved by the Institutional Review Board (IRB) of Severance Hospital (IRB 6-2004-1024).
A total of 54 patients lost HBsAg during follow up. Liver biopsy specimens and serum samples were available from 25 of them for further analysis. Paraffin embedded blocks of liver biopsy specimens were obtained from 20 patients following HBsAg loss, while refrigerated biopsy tissues were obtained from the remaining patients (No. 21 to 25). For three individuals (No. 2, 5, 6) liver specimens were also available before HBsAg loss. Twenty-eight HBsAg-positive patients served as controls.
DNA extraction from serum or liver tissues
DNA was extracted from the biopsy tissues or serum samples. Refrigerated tissue samples were digested with protenase K (2 mg/ml) at 37℃ for 1 hour and DNA was extracted using DNA Blood Mini Kit (Qiagen, Mildren, Germany) according to the manufacturer's instruction. DNA was eluted in 100 µL or 25 µL of double-distilled water, quantified by spectrophotometry at 260 nm and stored at -20℃ before use. Paraffin embedded tissue samples were deparaffinized with xylene and ethanol. Rehydrated liver tissues were treated with proteinase K and DNA was extracted with DNA Blood Mini Kit.
Sequencing of MHR and generation of HBsAg expressing constructs
The S gene sequence encoding MHR was amplified from DNA extracted from paraffin embedded tissue blocks using 0.5 µM each of sense primer (5'-GTCCTCTAATTCCAGGATC-3'), antisense primer (5'-GATGCTGTACAGACTTGG-3'), 3 ml of DNA template, 0.25 mM of dNTPs, and 0.5 U of Taq DNA polymerase (Takara, OTSU, Japan). The GeneAmp PCR System 9600 (Perkin Elmer, Norwalk, USA) was used and 40 cycles of amplification were performed. The PCR products were cloned into the pGEM-T easy vector (Promega, Madison, USA). Plasmid DNA was prepared by DNA midi kit (Qiagen, Valencia, USA) and sequenced with an automatic sequence analyzer. To amplify the entire HBV surface gene, the primer sets used were sense: 5'-TCGGAATTCTCAAATGTATACCCAAAGACAAAAGAA-3' and antisense: 5'-GGTAAGCTTATGGAGAACACAACATCAGGA-3'. Following cloning into pGEM-T vector, the HBV insert was released by double digestion with EcoRI and HindIII, purified by gel extraction and subcloned to pcDNA3.1, a mammalian expression vector (Invitrogen, Groningen, Netherlands). The wild-type ayw (pSV2A-Neo-(HBV)2)20 and adr (kindly provided by Dr. Ryu) strains were also cloned to the same vector. All clones were sequenced. Phylogenetic analysis was performed by using web-based Phylogeny.fr (http://phylogeny.lirmm.fr/phylo_cgi/simple_phylogeny.cgi).21
Transfection and detection of HBsAg
HepG2 cells seeded overnight in 6-well plates to 50-70% confluency were transfected with the surface gene constructs mentioned above using Lipofectamine Plus (Gibco-BRL). Transfection efficiency, as analyzed by flow cytometric analysis of GFP expression, was about 50 %. Culture media were collected at 24 hours post-transfection. Cells were detached by trypsin, and treated with lysis buffer (25 mM Tris/HCl, pH 7.4, 1% Triton X-100, 150 mM NaCl, 5% EDTA, 10 mM NaF, 1 mM phenylmethylsulfonyl fluoride (PMSF), and 10 µg/mL of aprotinin and leupeptin) for 30 min on ice, with vigorous vortex from time to time. The cell lysate was clarified by centrifugation for 10 min at 12,000 rpm, and supernatant was collected. Media and cell lysates were tested by ELISA for the presence of HBsAg using MonoLisa Ag HBs Plus (Biorad, Marnes la Coquette, France). Plates were read at a wavelength of 450 nm.
In Vitro transcription and translation assay
The TnT Quick Coupled Transcription and Translation system (Promega, Madison, USA) was used to generate [35S]-labeled HBsAg protein from the S gene cloned in pcDNA3.1 vector, using the T7 promoter present on the vector.20 Briefly, rabbit reticulocyte lysate was mixed with 1 mg of plasmid DNA, 2 µL of [35S]-methionine (Amersham, 1,000 Ci/mmol at 10 mCi/mL) and nuclease-free water to a final volume of 50 mL. The reaction mixture was incubated for 90 min at 30 ℃. Expression of each construct was determined by 12 % SDS-PAGE. After fixation for 30 min, the gel was treated with Amplify solution (Amersham, Little Chalfont, UK) and the radio-labeled protein was detected by exposure to X-ray film.
Quantification of intrahepatic HBV ccc DNA using Real-time PCR
For the detection of intrahepatic HBV ccc DNA, PCR was performed with 2.5 µL of DNA template, 0.5 µM of primers (sense: 5'-CTCTACCGTCCCCTTCTTCATCTGC-3'; antisense: 5'-CTTTATACGGGTCAATGTCCA-3'), 0.25 mM dNTPs, and 0.5 U of Taq polymerase. The primers span the DR2 and DR1 sites of HBV genome. The temperature conditions were as follows: 1 cycle of 95 ℃ for 4 min, 32 cycles of 95 ℃ for 45 s, 55 ℃ for 1 min, and 72 ℃ for 45 s. The PCR products were purified by gel extraction and sequenced with automatic sequencing analyzer for the mutation analysis of precore/core region.
The levels of intrahepatic HBV ccc DNA were quantified by the real-time PCR method as described previously,22 with 2.5 mL of template, 250 nM of the TaqMan probe (5'-FAM-TTCAAGCCTCCAAGCTGTGCCTTG-TAMRA-3') and 900 nM of the two PCR primers (sense: 5'-ACTCTTGGACTCBCAGCAATG-3' and antisense: 5'-CTTTATACGGGTCAATGTCCA-3'). The PCR was carried out in a 25ml volume using TaqMan Universal PCR Master Mix (Applied Biosystems, Branchburg, USA), with the following cycling conditions: 1 cycle of 95 ℃ 10 min, and 45 cycles of 95 ℃ for 30 s and 61.5 ℃ for 1 min. The GeneAmp 5700 Sequence Detection System (Applied Biosystems, Foster, USA) was used. Serial dilution of plasmid containing the HBV genome of ayw (GenGank no, J02203) was used as standards for HBV ccc DNA quantification. The content of intrahepatic HBV ccc DNA was derived by interpolation of the Ct value with the standard curve obtained from standard DNA ranging from 0 to 107 copies.
RESULTS
Detection of HBV DNA from serum and liver samples of HBsAg loss patients
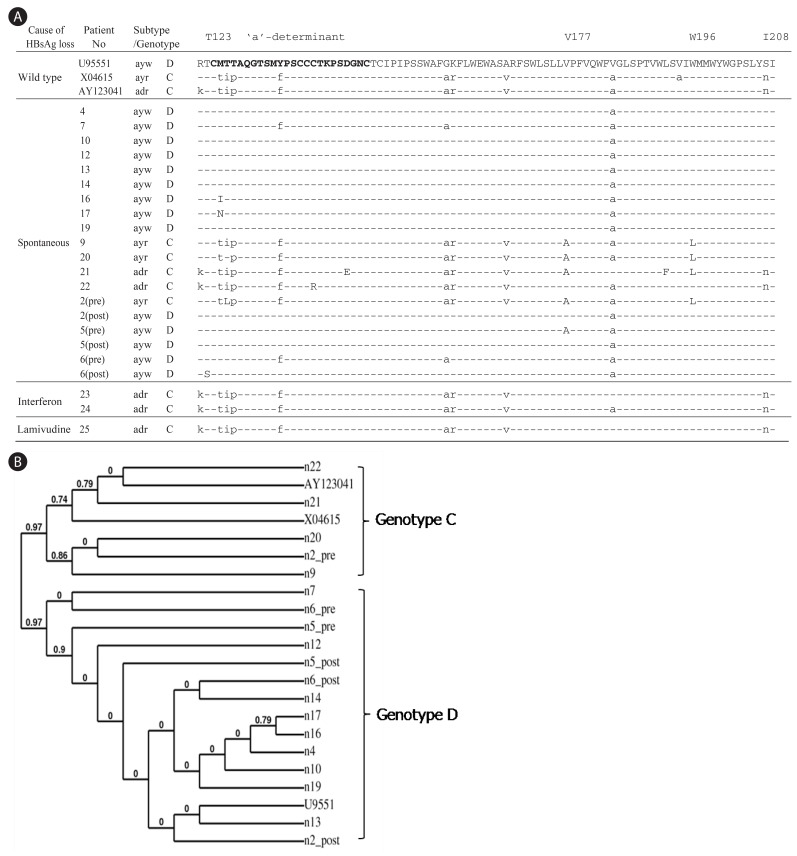
From a total of 435 patients with chronic hepatitis, liver cirrhosis, or hepatocellular carcinoma, 54 individuals lost serum HBsAg during the follow up. Among them, liver biopsies and serum samples were available from 25 cases at the HBsAg negative stage for further analysis. Table 1 summarizes their clinical data. Whereas the MHR region of HBV genome was undetectable from serum or biopsy samples of the 20 patients (No. 1-20) following 32 cycles of PCR amplification, a faint band of expected size was visible in agarose gels from all samples following 40 cycles of amplification and use of excess amount of template DNA. Due to the low yield of PCR product, screen of every 60-70 colonies by restriction enzyme digestion of miniprep DNA routinely yielded only one recombinant clone. Consequently, cloning of the MHR region was unsuccessful for 6 of the 25 patients. For the remaining 19 patients, one clone was obtained from each patient. For patients 2, 5, and 6, a clone derived from the HBsAg positive stage was obtained for comparison (Fig. 1). Cloning of the entire surface gene from tissue blocks was impossible from these 20 patients since not enough DNA was available from the thin paraffin sections. On the other hand, the fresh liver tissues from patients 21-25 allowed us to amplify and clone the entire surface gene.
Pattern of amino acid changes in the surface gene
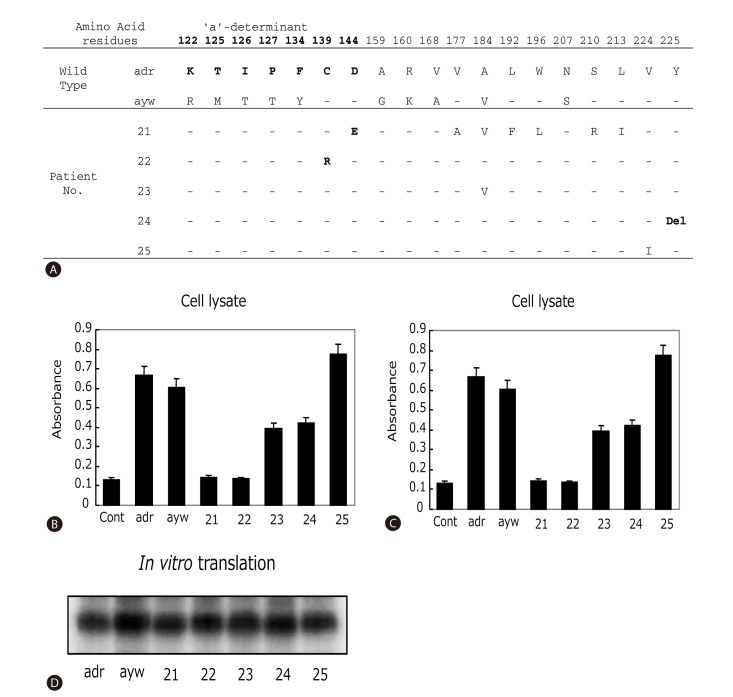
The cloned MHR region was sequenced. Four out of 19 patients (21%) harbored rare mutations inside the 'a'-determinant: M125I, M125N, C139R, and D144E (Fig. 1A). Taking the entire MHR region into consideration, three additional patients contained amino acid substitutions: T123S, V177A, L192F, and W196L. Of the three individuals for whom HBV sequences before and after HBsAg loss were available, only one patient had a specific mutation after HBsAg loss (T123S, patient 6). Interestingly, patient 2 displayed subtype change from ayr to ayw after the HBsAg loss. Deletion of residue 225 in the C-terminus of HBsAg (Y225del) was found in an interferon-treated patient (Fig. 2A).
Relationship between spontaneous HBsAg loss and HBV subtypes in Korea
Previous reports demonstrated subtype adr (genotype C) as the dominant HBV in Korea, accounting for more than 90 % of infection.23,24 To establish the association between spontaneous HBsAg loss and viral genotypes, the HBV subtype and genotype in each patient was deduced from the sequence profile in the MHR region as described.25 The HBV genotypes were also verified by phylogenetic analysis21 (Fig. 1B) and RFMP analysis (data not shown). Unexpectedly, 75 % (12 of 16) of patients who lost HBsAg spontaneously were seemed to be infected with the ayw subtype with sequence similar to genotype D (Table 2). Two patients (12.5 %) were infected with subtype ayr (genotype C) and the remaining two (12.5%) with adr (genotype C). To exclude the possibility that the preponderance of genotype D in this study reflects regional difference in the distribution of HBV genotypes in Korea, we cloned and sequenced the whole surface gene from 28 HBsAg-positive patients enrolled in our hospital. As a result, 27 of the 28 HBsAg positive patients (96%) were infected with adr subtype (genotype C) (Table 2). The remaining patient was infected with adw subtype (genotype A). Therefore, our results suggest a much higher rate of genotype D to clear HBsAg than genotype C in Korea. Alternatively, genotype D may have strong selective advantage against genotype C at the HBsAg negative stage of infection.
Mutations found in HBsAg negative patients reduced HBsAg affinity to its antibody or impaired its secretion
To investigate the contribution of mutations in the surface gene on HBsAg levels, surface genes from patients 21-25 were cloned into pcDNA3.1 vector and expressed either in reticulocyte lysate as radio-labeled protein or in HepG2 cells. Protein expression levels in the cell-free system as measured by autoradiography of the SDS-PAGE were comparable among the clones and in comparison with the wild-type controls (Fig. 2D). However, when assayed from culture supernatant of transfected HepG2 cells using anti-HBs antibody (ELISA assay), The HBsAg levels were much lower for clones derived from patients 21, 22, and 24 (Fig. 2C). Analysis of HepG2 cell lysate revealed similar low levels of HBsAg for clones 21 and 22, but near normal levels for the clone 24 (Fig. 2B). Together, these observations suggest that mutations present in clones 21 and 22 impaired HBsAg affinity to its antibody, while mutations found in clone 24 blocked HBsAg secretion. An inspection of HBsAg sequences shown in Fig. 2A suggests that C139R (clone 22) and D144E (clone 21) are the likely mutations responsible for impaired HBsAg recognition, whereas deletion of Y225 (clone 24) may have reduced HBsAg secretion. Finally, patient 23 and 25, with interferon and lamivudine-mediated HBsAg loss, respectively, showed no gross reduction in HBsAg levels in cell lysate or culture medium.
Quantification of intrahepatic HBV ccc DNA and amino acid changes in precore/core region
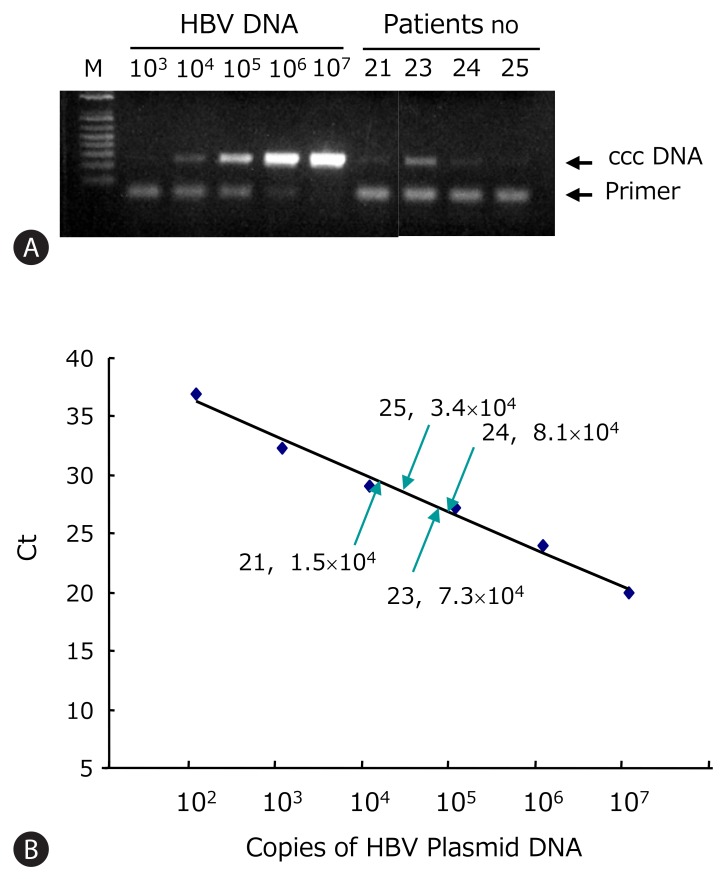
HBV ccc DNA from patients 21-25 was detected using the PCR primers flanking the direct repeat region (Fig. 3A). All showed detectable ccc DNA by PCR. These PCR products were verified by nested PCR and direct sequencing. The A1762T and G1764A core promoter mutations were found only in the lamivudine-treated patient. The Sp1 binding sites were mutated in clones 21, 22, and 25 (data not shown). Using the real-time PCR, the intrahepatic HBV ccc DNA was quantified. This assay allowed linear quantification of HBV ccc DNA in the range 102-107 copies/reaction of standard plasmid (Fig. 3B). The ccc DNA copies were normalized per microgram of liver DNA. The concentration of viral genomes in the HBsAg-negative liver tissues varied from 104 to 107 copies/mg of liver DNA which corresponds to 0.6 to 8.0 copies of ccc DNA per cell (Table 3).
DISCUSSION
In the present study, we characterized HBV genomes from patients who lost HBsAg spontaneously or following antiviral therapy. Hepatitis B surface antigen possesses two pairs of group-specific determinants permitting the classification of all HBV strains into four major serological subtypes: adr, adw, ayr and ayw. Although many studies have reported presence of HBV DNA in HBsAg negative patients,1,2,3,4,5,6,7,8,9,10,11,12,13,14,15 only a few reports examined role of HBV subtypes26 or genotypes.37 Furusyo et al26 found that Japanese carriers of adr subtype had an increased probability of clearing HBsAg compared with those infected with adw subtype. However, with an enzyme immunoassay for subtype determination they were unable to confirm viral subtype at the HBsAg negative stage of infection. In this work, we found that 75 % of spontaneous HBsAg loss patients in Korea seemed to harbor subtype ayw (genotype D) at the HBsAg negative stage (Table 2). Only 25% were infected with genotype C (12.5 % with subtype ayr, and 12.5% with subtype adr), the predominant HBV genotype in Korea accounting for more than 90 % of infection. Therefore, our findings suggest a possible link between the spontaneous loss of HBsAg and a particular HBV subtype, ayw, in Korea. From the three individuals for whom samples before and after HBsAg loss were available, two were infected with genotype D, while the third (patient 2) showed genotype change from C to D (Fig. 1). Therefore infection with genotype D may be more predisposed to HBsAg loss, or genotype D is selected following HBsAg loss in patients with mixed infection with genotype C. In this regard, Bahn and coworkers found genotype switch from A to D in three pediatric patients following seroconversion from HBsAg to anti-HBs.27 A large-scale prospective study is needed to clarify this issue.
Only 7 of 19 (36.8 %) patients harbored mutations in the MHR, which is similar to 43 % of mutation rate found in HBsAg-negative patients in China.10 Therefore, factors other than mutated MHR region may contribute to occult HBV infection. Functional analysis through transfection experiments in HepG2 cells suggest that some of the mutations found in our patients do impair HBsAg antigenicity (C139R, D144E) or block its secretion (Y225del). There are eight highly conserved cysteine residues in the MHR of HBsAg. Of these, residues 107, 137, 138, 139, 147 and 149 are suggested to stabilize the conformation of HBsAg by formation the intramolecular disulfide bridges.28,29 Therefore, the C139R mutation observed in patient 22 may destroy the major antigenic determinant thus reducing affinity to anti-HBs (Fig. 2). Mutations at positions 144 and 145 of the 'a'-determinant of HBsAg are commonly found in liver transplant recipients with failed hepatitis B immune globulin prophylaxis.30,31,32,33,34 A mixture of D144G/D144A or combined D144E / G145R mutations were detected after hepatitis B immune globulin treatment. We previously reported that surface gene isolated from a liver transplantation patient receiving hepatitis B immune globulin displayed reduced HBsAg antigenicity in transfection experiments.34 This mutant surface gene shares with clone 21 the D144E mutation. Thus, we are confident that this point mutation was responsible for impaired HBsAg antigenicity in clone 21.
Very little is known about the molecular basis of HBsAg secretion defect. In a previous report, a combination of amino acid changes in the surface protein, including G145R, was found to cause secretion defect of HBsAg.35 Clone 24 contained no mutations in the surface protein except deletion of the penultimate residue, Y225 (Fig. 2A). Transfection results revealed a specific secretion defect of clone 24, suggesting that this novel amino acid deletion could partly explain the HBsAg loss in this particular patient. Whether this deletion would also impair virion secretion is presently unknown. In this regard, we previously found several point mutations in the MHR region36 and W172stop corresponding to the multidrug-resistant rtA181T mutation in polymerase37 impair the secretion of both virions and HBsAg.
One major reason for the loss of HBsAg in our patients could be reduced viral gene expression and genome replication, which is characteristic of patients negative for HBsAg. This concept is consistent with the observation that 40 cycles of PCR were required to amplify HBV DNA from all the patients. The ccc DNA is the template for viral gene transcription and genome replication. We found 104-107 copies of ccc DNA /mg of liver DNA in Korean HBsAg negative patients (Fig. 3). In contrast, Zanella et al38 reported 102-107 of HBV genomes/mg of liver DNA from HBsAg-positive patients and 101-104 genomes/mg of liver DNA from HBsAg-negative patients. The reason for the discrepancy is not clear. At any rate, loss of HBsAg in Korean HBV carriers is characterized by low levels of ccc DNA, infection with genotype D (serotype ayw), or in certain cases, presence of mutations in the HBsAg gene that impair HBsAg antigenicity or secretion.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print