The mutational landscape of hepatocellular carcinoma
Article information
Abstract
The development of hepatocellular carcinoma (HCC) is a complex process, and HCC arises from the accumulation of multiple genetic alterations leading to changes in the genomic landscape. Current advances in genomic technologies have revolutionized the search for genetic alterations in cancer genomes. Recent studies in which all coding exons in HCC were sequenced have shed new light on the genomic landscape of this malignant disease. Catalogues of these somatic mutations and systematic analysis of catalogued mutations will lead us to uncover candidate HCC driver genes, although further functional validation is needed to determine whether these genes play a causal role in the development of HCC. This review provides an overview of previously known oncogenes and new oncogene candidates in HCC that were uncovered from recent exome or whole-genome sequencing studies. This knowledge provides direction for future personalized treatment approaches for patients with HCC.
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common cancers in the world, accounting for an estimated 600,000 deaths annually.1 HCC is common in Southeast Asia and sub-Saharan Africa, but the incidence rate of HCC has also increased in the United States and Western Europe over the past 25 years, and incidence and mortality rates of HCC are likely to double over the next 10 to 20 years.234
Although much is known about both the cellular changes that lead to HCC and the etiologic agents responsible for most cases of HCC (i.e., hepatitis B and C viral infections and alcohol abuse), the molecular pathogenesis of HCC is not well understood.567 Moreover, the severity of HCC, the lack of good diagnostic markers and treatment strategies, and clinical heterogeneity make management of the disease a major challenge.78
Patients with HCC have a highly variable clinical course,69 indicating that HCC comprises several biologically distinctive subgroups. Despite the considerable efforts that have been devoted to establishing a classification system,691011121314 clinical and pathologic diagnosis and classification of HCC remain unreliable in predicting patient prognosis and response to therapy. The prognostic variability likely reflects a molecular heterogeneity that has not been identified from methods traditionally used to characterize HCC. Improving the classification of HCC to more accurately predict patient prognosis and clarify the underlying biology of HCC development at the molecular level would improve the application of currently available treatment modalities and offer new potential treatment strategies.
The most exciting cancer research developments in recent years have involved the clinical validation of molecularly targeted drugs that inhibit the action of pathogenic gene products such as protein kinases and proteinase.1516 Treatment with these targeted drugs can more efficiently alter the natural history of disease and reduce mortality. Identification of cancer type-specific oncogenes that play a key role in the progression of a certain cancer can lead to advances in the classification of the cancer and development of molecularly targeted therapies. However, characterization of HCC at the molecular level to identify altered oncogenes (i.e., potential therapeutic targets) has lagged compared with other cancers. Therefore, to improve treatment options and reduce mortality, we must develop treatment strategies that can be applied in the near future while improving our understanding of hepatocarcinogenesis. This review discusses recent studies of cancer genomics in HCC aimed at improving understanding of the molecular pathogenesis of the disease.
Sequencing technologies in genomics
Growing demand for more efficient DNA sequencing has given rise to new technologies. Next-generation sequencing (NGS) in general refers to second-generation sequencing technologies that are more cost-efficient and have higher throughput than the first-generation Sanger sequencing. NGS was developed from new technologies and computing systems that can control complex and big data. NGS includes various sequencing methods to read a large volume of sequence quickly and inexpensively. Many different companies have developed various methods, such as the 454 Genome Sequencer FLX System (Roche Applied Science, Indianapolis, IN, USA), Sequencing by Oligonucleotide Ligation and Detection or SOLiD (Life Technologies, Carlsbad, CA, USA), HiSeq X (Illumina, San Diego, CA, USA), HeliScope Single Molecule Sequencer (Helicos BioSciences, Cambridge, MA, USA), Single Molecule Real Time or SMRT sequencing (Pacific Bioscience, Menlo Park, CA, USA), and the Ion Proton System (developed by Ion Torrent Systems, now owned by Life Technologies). Faster and more cost-effective sequencing methods are still under development. These new technologies provide unique opportunities to investigate all sequences of entire cancer genomes and to understand how genetic differences affect disease.
Depending on the target sample resource and coverage, NGS approaches can include whole-genome sequencing, whole-exome sequencing, or whole-transcriptome sequencing. NGS is challenged by massive genomic data, including considerable unknown information that may be valuable or simply represent nonsense mutations. As more data are accumulating and innovative analytic methods are developing, NGS is becoming a key tool for elucidating key oncogenic pathways among the many heterogeneous genetic aberrations in HCC.
Cataloguing somatic alteration in HCC
To gain a comprehensive view of the genetic alterations underlying HCC, many investigators have analyzed the HCC genome using NGS technologies. In the first comprehensive analysis of the HCC genome, the researchers sequenced the entire genome of hepatitis C virus-positive HCC tissue and uncovered many previously unrecognized mutations.17 In a later study, researchers sequenced exons of 18,000 protein-coding genes (exome) in 10 HCC tumors and discovered that the AT-rich interactive domain 2 (ARID2) gene was frequently mutated.18 Parallel to these studies, Tao et al analyzed whole-genome sequencing data from primary and recurrent HCC tumors from the same patient to monitor the evolution of the HCC genome during clinical progression.19 More comprehensive analyses of the HCC genome followed, in which researchers sequenced multiple HCC genomes and uncovered candidate driver genes.20212223 In addition, whole-genome sequencing of 88 hepatitis B virus-related HCC tumors uncovered many candidate driver mutations largely driven by hepatitis B virus integrations.24
More recently, several teams of investigators completed a large-scale analysis of the genetic makeup of HCC.252627282930 These studies each examined a large number of HCC tumors (42 to 503 tumors) and uncovered many somatic mutations frequently observed in HCC.
Frequently mutated genes in HCC
Since the first genomic sequencing studies for HCC were published,1718 many other studies have catalogued potential driver genes in HCC. Table 1 summarizes the frequently mutated genes identified in large-scale studies that examined 40 or more HCC tissue samples.252627282930

Genes most frequently mutated in hepatocellular carcinoma, identified in large-scale studies
TERT, regulating telomere length
Telomerase reverse transcriptase (TERT) encodes a rate-limiting catalytic subunit of telomerase that is essential to maintain telomere length and plays a pivotal role in stem cells, aging, and cancer.3132 Expression of TERT is mostly repressed in somatic cells except for self-renewing cells such as stem cells.33 In contrast, 70-90% of cancer cells stably express this enzyme, which is reactivated during tumorigenesis and is necessary for unlimited proliferation of cancer cells.343536
The human TERT gene is located on chromosome 5p15. Transcriptional regulation is considered to be the major mechanism of telomerase regulation. The TERT promoter contains a GC-rich sequence lacking the typical TATA box sequence.373839 The core promoter region consists of about 260 base pairs with multiple transcription factor binding sites, including E-boxes, where MYC binds.40 Sp1 is also a key transcription factor that binds to GC-rich sites on the core promoter and activates TERT transcription.41 Full activation of the TERT promoter requires cooperative action of MYC and Sp1.
The precise mechanism behind TERT activation in cancers mostly remains unknown. However, the newly described recurrent somatic mutations in the TERT promoter in melanoma and other cancers has provided novel insight into the possible cause of tumor-specific increased TERT expression.424344 In particular, TERT promoters have been found to be mutated in more than 50% of HCC tissue samples examined, making them the most frequently occurring single-nucleotide mutations observed in HCC.3045 These mutations create a potential binding site for E-twenty six/ternary complex factors (ETS/TCF) transcription factors and are predicted to increase promoter activity and expression of TERT. Indeed, a recent study further demonstrated that a specific transcription factor called GABP, a member of the ETS family, is selectively recruited to the mutated form of the TERT promoter and activates TERT expression.46
TP53, regulating the cell cycle and genomic integrity
Tumor protein 53 (TP53) is the second most frequently mutated gene in HCC, occurring in more than 30% of cases of HCC.25304748 TP53 functions as a tumor suppressor gene by initiating cell-cycle arrest, apoptosis, and senescence in response to several cellular stresses, including DNA damage, oncogene activation, and hypoxia, to maintain the integrity of the genome.495051 TP53 protein acts as a transcription factor that binds to specific DNA sequences as a tetramer and is generally regulated by the MDM2/E3 ubiquitin ligase that interacts with the TP53 transactivation domain, blocking p53-mediated transcriptional activity and promoting proteasome-dependent TP53 degradation at the same time. As a transcription factor, TP53 can both activate and repress gene expression, controlling expression of genes involved in cell cycle arrest, apoptosis, and senescence. However, recent studies demonstrated that TP53 also plays important roles in cellular metabolism, autophagy, oxidative stress, and stem cell maintenance.52
Most mutations of TP53 found in HCC are missense mutations that reside in the DNA-binding domain of TP53, resulting in a lower affinity to bind the sequence-specific response elements of TP53 target genes (Fig. 1). Although most mutations in TP53 result in loss of function, some mutations give rise to novel oncogenic activities that are independent of wild-type TP53 (gain-of-function mutations), such as angiogenesis, metastasis, and resistance to standard therapies.5253
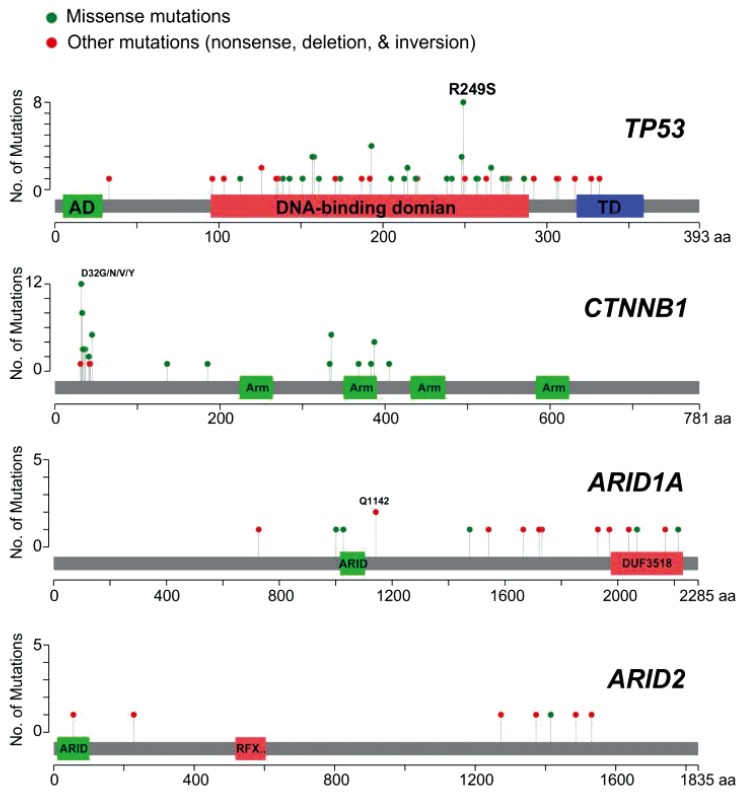
Distribution of somatic mutations in frequently mutated genes in hepatocellular carcinoma. Mutation data were obtained from The Cancer Genome Atlas project hepatocellular carcinoma samples (n=193).
A recent study demonstrated that the TP53 mutations are associated with poor prognosis in HCC.47 In particular, hotspot mutations such as R249S and V157F are strongly associated with poor prognosis, indicating that these mutations can be used as prognostic markers in HCC.
CTNNB1 and AXIN1, regulating the WNT pathway
Catenin beta 1 (CTNNB1) encodes β-catenin, which is a subunit of the cadherin protein complex on the cellular surface that acts as a signaling molecule in the wingless-type MMTV integration site family (WNT) pathway.5455 When WNT signaling is absent, cytosolic β-catenin protein levels are low because of phosphorylation-dependent ubiquitination and degradation that is orchestrated by the axis inhibitor (AXIN) complex, which is composed of AXIN, APC, CK1, and GSK3. When WNT ligands bind to a Frizzled receptor and its co-receptor, LRP5/6, WNT induces a receptor complex formation, which relocates AXIN to the plasma membrane, resulting in stabilization of β-catenin. Stabilized β-catenin forms a complex with the ternary complex factor/lymphoid enhancer factor (TCF/LEF) and increases transcription of genes involved in cell growth.
CTNNB1 is one of most frequently mutated genes in HCC. Aberrant activation of β-catenin has been observed in 20-30% of HCC patients.213056 Most mutated residues are phosphorylation sites or near to phosphorylation sites,21305657 preventing phosphorylation of β-catenin (Fig. 1). Thus, β-catenin is constitutively activated by mutations. Interestingly, previous studies have shown that mutations in β-catenin are almost mutually exclusive with mutations in TP53.2558 These observations strongly suggest that HCC with a β-catenin mutation may represent a clinically distinct subtype of HCC. Mutations in the WNT pathway also have been reported in the degradation complex. AXIN1 is the second most frequently mutated gene in the WNT pathway (occurring in 5-10% of cases).25293059
Interestingly, activated mutations of CTNNB1 are most significantly associated with mutations in TERT promoters,3045 suggesting potential interaction between these 2 genes in hepatocarcinogenesis. This is further supported by a recent observation that mutations in CTNNB1 and TERT promoter are key alterations occurring early and late in the transition from adenoma to carcinoma in the liver.58 Furthermore, recent studies demonstrated that TERT is a direct target of CTNNB1, in cooperation with KLF4, further supporting the idea that these 2 genes functionally interact in hepatocarcinogenesis.6061
ARID1A and ARID2, remodeling chromosomes
ARID1A and ARID2 are also frequently mutated in HCC (in up to 20% of cases).182930 They belong to the AT-rich interaction domain (ARID) family, which contains 7 subfamilies and 15 members; ARID genes are characterized by a 100-amino acid DNA-binding ARID domain.62 ARID1A associates with several other proteins to form the BRG1-associated factor (BAF) complexes, a subfamily of a switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex.63 This complex uses the energy from ATP to mobilize nucleosomes by sliding, ejecting, and inserting histone octamers, thereby regulating DNA accessibility to other cellular machineries involved in transcription, DNA replication, and repair. Following the discovery of mutations in ARID1A in gynecologic cancers, including ovarian, endometrial, and uterine cancers,64656667 mutations in ARID1A were found in many other cancers, including breast cancer, esophageal cancer, gastric cancer, pancreatic cancer, bladder cancer, and prostate cancer.6268
Most cancer-associated mutations in ARID1A appear to be loss-of-function mutations; nonsense or frameshift rather than missense mutations in ARID1A are the dominant forms in many cancers, including HCC (Fig. 1), suggesting that ARID1A is a tumor suppressor. In a recent study, ARID1A knockdown significantly increased cell growth in wild-type HCC cell lines but had no effect in a cell line with an ARID1A mutation,22 further supporting the idea that ARID1A functions as tumor suppressor gene in HCC.
ARID2 is a member of the polybromo-associated BRG1-associated factor (PBAF) complex, another SWI/SNF complex involved in ligand-dependent transcriptional activation by nuclear receptors.63 Although mutations in ARID2 are less common than those in ARID1A, most mutations in ARID2 are also loss-of-function mutations, as seen in ARID1A (Fig. 1).
Interestingly, recent studies showed that ARID1A mutations are negatively associated with mutations in TP53 in gastric cancer,6970 indicating that ARID1A and TP53 may work together in codependent manner to suppress tumor development. Analysis of HCC mutation data from The Cancer Genome Atlas project also showed that mutations in ARID1A/ARID2 and were negatively associated with mutations in TP53, further supporting the idea that these 2 genes interact (Fig. 2). However, it remains to be determined how ARID1A interacts with TP53 or whether ARID1A or ARID2 can modulate TP53 activity.
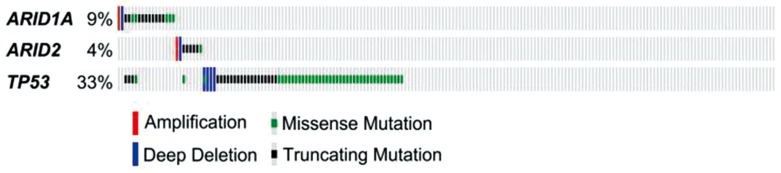
Genetic alterations of ARID1A, ARID2, and TP53 in hepatocellular carcinoma. Mutation data were obtained from The Cancer Genome Atlas project hepatocellular carcinoma samples (n=193), which are displayed as columns.
NFE2L2 and KEAP1, regulating the oxidative stress pathway
Reactive oxygen species (ROS) are the byproduct of many different cellular activities and are well recognized for their role in various diseases, including cancer. ROS can interact with and damage DNA, RNA, and proteins, resulting in spontaneous mutations leading to the initiation of many cancers.71 Nuclear factor erythroid 2-like 2 (NFE2L2), also known as NRF2, is a member of the cap 'n' collar (CNC) family of basic region leucine zipper transcription factors and a key regulator of important signaling pathways involved in cellular defense and survival against oxidative stress.727374 Under normal physiologic conditions, intracellular levels of NFE2L2 are kept low by its cytosolic inhibitor, Kelch-like ECH-associated protein 1 (KEAP1), which functions as an adaptor protein in the cullin 3-based E3 ligase complex that ubiquitinates NFE2L2 in the cytoplasm and targets it for proteasomal degradation. However, under oxidative stress, the activity of KEAP1 is diminished and ubiquitination of NFE2L2 is disturbed, allowing nuclear accumulation of NFE2L2. In the nucleus, NFE2L2 binds to the antioxidant response element in the regulatory regions of the target genes and drives expression of these target genes.
Because of the known molecular activity of NFE2L2, in particular in preventing DNA damage and mutagenic events, NFE2L2 has been recognized as a tumor inhibitor for a long time, thus providing a rationale for developing tumor-prevention strategies using NFE2L2 activators. However, this view was changed by the finding that NFE2L2 has a protective role not only in normal cells but also in cancer cells, in which aberrant signaling via the KEAP1-NFE2L2 pathway leads to constitutive activation of NFE2L2 and upregulation of its target genes, resulting in enhanced survival of cancer cells.757677
NFE2L2 consists of 7 highly conserved domains called Neh1 to Neh7 (Nrf2-ECH homology). While Neh1 contains the CNC-bZIP structure, promoting dimerization with its other partner MAF proteins and binding to DNA, the Neh2 domain possesses DLG and ETGE motifs, which play an important role in negative regulation by binding to KEAP1.78 Mutation rates of KEAP1 and NFE2L2 in HCC are around 3-5%.262930 Importantly, mutations in NFE2L2 are clustered around amino acid residues in the DLG and ETGE motifs, which are negative regulatory sites, strongly indicating that mutations lead to constitutive activation of NFE2L2 in HCC. Consistent with this, most mutations in KEAP1 are loss-of-function mutations, also leading to constitutive activation of NFE2L2.
During tumor promotion, high cellular activity generates an increased ROS burden that causes cell cycle arrest or apoptosis. Therefore, it is highly likely that upregulation or constitutive activation of NFE2L2 would provide significant benefits to cancer cells. Recent studies also showed that activation of NFE2L2 is significantly associated with poor prognosis as a result of constitutive expression of cytoprotective genes in cancer cells,7679 further supporting the idea that NFE2L2 plays a cytoprotective role in cancer cells. Therefore, it is suggested that once tumor formation is initiated, cancer cells hijack the NFE2L2-KEAP1 system to acquire stress resistance and a growth advantage.
Future direction: personalized medicine
Many genome-wide studies of HCC have clearly demonstrated that HCC is a genetically heterogeneous disease. The more data that are collected from HCC genomes, the more evident it becomes that each tumor has its own set of genetic alterations. Better understanding of such genetic alterations in cancer cells improves diagnosis, prognosis, and treatment of HCC, which can be based on the specific molecular alterations that drive individual tumors.
Personalized medicine is a phrase that is often used to describe an innovative approach that takes into account personal differences such as genetic makeups, environments, and lifestyles. Determining the exact genetic alterations driving tumor development and providing definite molecular diagnoses are core elements of personalized medicine. Current cancer genomics research aims to advance personalized medicine by collecting and analyzing data from cancer genomes to uncover novel genetic or epigenetic alterations associated with specific subtypes of cancers. This gives clinicians the resources they need to develop more effective ways to diagnose, treat, and prevent cancer.
Such genomic information has already helped to re-shape clinical practice surrounding cancer treatments. For example, treatment with trastuzumab was found to significantly improve outcomes and prolong survival in breast cancer patients with HER2-positive disease, and today the drug is established as the standard of care.8081 Other studies have shown that lung cancer patients with EGFR mutations respond best to drugs targeting these mutated receptors, such as gefitinib and erlotinib.8283 In contrast, patients with KRAS-mutated colon tumors had little or no response to drugs targeting the EGFR pathway.8485
In the near future, it is anticipated that tumors in HCC patients will be systematically surveyed to identify the underlying somatic genetic changes in sequence, expression, and copy number and patients will be treated according to the genetic or epigenetic makeup of the HCC cells. However, most genetic alterations observed in HCC are associated with a loss of function. Actionable target genes found in other cancers, such as those encoding protein kinases and proteins with enzymatic activity, are not significantly mutated in HCC. Furthermore, frequently mutated genes in HCC such as CTNNB1 and ARID1A/2 are not actionable targets yet. Therefore, alternative approaches must be devised to find actionable targets by systematically integrating multiple genetic, epigenetic, and proteomic data from HCC tumors, as demonstrated by the success of The Cancer Genome Atlas projects in many cancers.8687888990919293
Acknowledgements
The study was supported in part by the 2011 and 2012 cycles of the MD Anderson Sister Institute Network Fund.
Notes
Conflicts of Interest: The author has no conflicts to disclose.
Abbreviations
ARID1A
AT-rich interactive domain 1A
ARID2
AT-rich interactive domain 2
AXIN1
axis inhibitor 1
CTNNB1
catenin (cadherin-associated protein) beta 1
HCC
hepatocellular carcinoma
KEAP1
Kelch-like ECH-associated protein 1
NFE2L2
nuclear factor erythroid 2-like 2
NGS
next-generation sequencing
TERT
telomerase reverse transcriptase
TP53
tumor protein 53
WNT
wingless-type MMTV integration site family