INTRODUCTION
Chronic hepatic inflammation which is caused by excessive alcoholic consumption, hepatitis B or C virus, and non-alcoholic steatohepatitis results in hepatic fibrosis.1 The terminal outcome of liver fibrosis is liver cirrhosis, characterized by excessive deposition of extracellular matrix proteins and the appearance of regenerative nodules, accompanied by hepatic failure, portal hypertention and hepatocellular carcinoma. Portal hypertension may cause serious complications such as esophageal variceal bleeding, ascites, and hepatic encephalopathy that are the major causes of death in cirrhotic patients. However, there is no effective anti-fibrotic therapy to treat or reverse hepatic fibrosis and liver cirrhosis. Therefore there is an urgent need to develop anti-fibrotic agent through the research for the molecular pathogenesis of hepatic fibrogenesis.
The hepatic stellate cell (HSC) is the main fibrogenic cell type in the injured liver and it also has been identified as an important effector in liver inflammation.1 HSCs are located in the space of Disse in close proximity to hepatocytes on one side and endothelial and Kupffer cells on the other side. Quiescent HSCs are desmin-positive, perisinusoidal cells that are the primary cell in the body responsible for vitamin A storage.1 Upon activation by liver injury, quiescent HSCs transdifferentiate into myofibroblast which produce inflammatory cytokines and several extracellular matrix proteins and glycoproteins including at least five collagen types, fibronectin, laminin, entactin, tenascin, and several proteoglycans. 2
Reactive oxygen species (ROS) are involved as key secondary messengers in numerous signaling pathways including transcriptional regulation, differentiation, proliferation to oncogenic transformation, and activation of programmed cell death.3 It has previously been demonstrated that ROS significantly contributes hepatic fibrogenesis from various liver injuries including alcohol abuse, hepatitis C virus infection, iron overload and chronic cholestasis.4,5 ROS can stimulate the production of the type I collagen,6 and may act as an intracellular signaling mediator of the fibrogenic action of TGF-╬▓.7,8 ROS may be generated by multiple sources including mitochondrial respiratory chain, cytochrome p4502E1, peroxisomes, and NADPH oxidases (NOXs) in the liver.5 Cumulating evidences indicate the critical role of NOX-mediated ROS in hepatic fibrogenesis which will be described in this review.
NADPH oxidase homologues and components
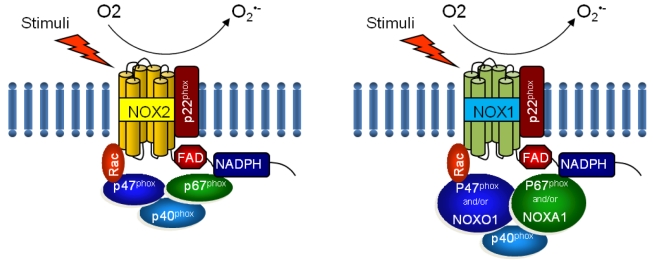
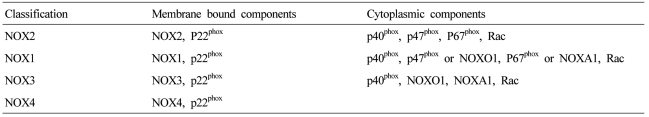
NOX is a multicomponent enzyme complex that generates ROS in response to a wide range of stimuli including TNF-╬▒, IL-1╬▓, leptin and angiotensin II (Ang II).9,10 NOX generates superoxide (O2┬Ę-) from molecular oxygen using NADPH as an electron donor, and superoxide converts to hydrogen peroxide (H2O2) by superoxide dismutase (SOD). Classic NOX is the phagocytic NOX found in neutrophils. The phagocytic NOX complex consists of the catalytic subunit gp91phox(renamed NOX2) together with the regulatory subunit p22phox located in the membrane. The other regulatory components p47phox, p40phox, p67phox and the small GTPase Rac are normally located in the cytoplasm.11 Upon stimulation with agonists, the cytosolic subunits translocate to the membrane-bound cytochrome complex leading to enzymatic activity. Humans have 4 additional homologous NOX proteins (NOX1, NOX3, NOX4, NOX5) and 2 related enzymes (DUOX1, DUOX2) that may function in non-phagocytes.11 Among the seven members of the NOX family, NOX1 is structurally and functionally similar to NOX2. While NOX1 is also p22phox-dependent, NOX1 may use NOXO1 (homologue of p47phox) to organize the enzyme assembly and NOXA1 (homologue of p67phox) for enzyme activation.12 In contrast, NOX4 requires only p22phox without recruitment of cytosolic regulatory components to exert its enzymatic activity.13 All NOX enzymes are able to catalyze the reduction of molecular oxygen to superoxide, but there are key differences in their activation, subunit composition, localization, and expression (Table 1, Fig. 1).14
NADPH oxidase-mediated ROS generation and human diseases
Chronic granulomatous disease (CGD) has been described for several decades as a human disease resulting from defects in NOX complex.15 The genetic defect of phagocytic NOX activity results in an inability to produce the superoxide anion necessary for killing bacteria and fungi by neutophils and phagocytes. The patients with CGD predispose to serious bacterial and fungal infections as well as granulomatous complications. It has been reported that at least 5 different genes involved in NOX activity may cause CGD.15 The gene mutation of NOX2 on the X chromosome account for about two thirds of the cases and present as severe form with earlier symptom manifestation. Meanwhile, mutations in p47phox, p67phox, and p22phox account for about one thirds of the cases as less severe form characterized by autosomal recessive inheritance.16
In contrast with CGD that are defective in NOX function, several human diseases are associated with excessive ROS production by an overactive NOX. NOX mediated oxidative stress may cause endothelial dysfunction, vascular smooth muscle contraction and hypertrophy that results in various vascular diseases such as hypertension, hypercholesterolemia, and atherosclerosis.14 Amyotrophic lateral sclerosis (ALS) is a progressive motor neuron degenerative disease. Although multiple factors seem to be implicated in the underlying pathogenesis, recent evidence strongly suggest that oxidative stress is an important component of disease progression of ALS.17 It was reported that oxidative damage is increased in both sporadic and familial form of ALS which is associated with mutations of SOD1.18 In a SOD1G93A transgenic mouse, an animal model of ALS, both NOX1 and NOX2 are involved in disease progression.19,20 In ALS pathogenesis, the activation of proinflammatory transcription factor NF-╬║B by TNF-╬▒ and IL-1╬▓ has been reported to involve NOX.17 More recently, the important role of neuronal inflammation associated with redox-active signaling endosomes (redoxosomes) has been reported.21 After TNF-╬▒ or IL-1╬▓ stimulation, dynamin-dependent co-endocytosis of the ligand-activated receptor and NOX complexes occur.22,23 As a next step, SOD1 is recruited to the redoxosome, where it binds to Rac1 and stabilizes the GTP-bound active form of Rac1.22 Oxidation of Rac1 by H2O2 uncouples SOD1 binding in a reversible fashion. The expression of ALS mutant forms of SOD1 (G93A) enhances NOX dependent superoxide production and Rac1 activation in redoxosome.24 Taken together, mutations in SOD1 leads to increased NOX dependent ROS production and hyperactivation of proinflamamtory signaling that can kill motor neurons to develop ALS, where endosomal NOX regulation by SOD1/Rac1 interaction seems to be critical.17 The possible disturbance of the above self-regulated redox signaling for NOX-derived ROS production may also be implicated in hepatic fibrogenesis. However, the detailed intracellular signaling of NOX-mediated superoxide production and subsequent conversion to H2O2 in HSCs is largely unknown.
Angiotensin II induces hepatic fibrosis via NADPH oxidase activation in hepatic stellate cells
Key elements of the renin angiotensin system such as angiotensin converting enzyme and angiotensin type I receptor (AT1R) are present in normal liver tissue, and there is major up-regulation of the system in the bile duct-ligated liver.25 Several studies demonstrated that inhibition of Angiotensin II (Ang II) synthesis and/or the blockade of AT1R markedly decrease inflammation and hepatic firosis in experimental models of chronic liver injury.26-28 In parallel, the study investigating the fibrogenic response in WT and AT1R-/- mouse demonstrated that hepatic collagen accumulation was lower in AT1R-/- mice compared to WT mice after bile duct ligation (BDL).29 p47phox-/- mice have been widely used as a genetic model to study NOX inhibition.30-32 Bataller et al. demonstrated that p47phox-/- mice showed attenuated liver injury and fibrosis compared with WT counterparts after BDL.33
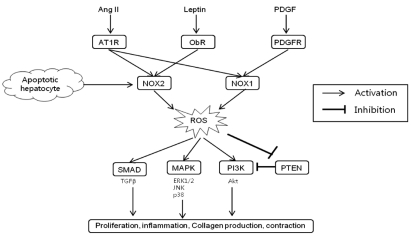
As shown previously in the kidney,34 Ang II exerts its activity for the regulation of intracellular pathways using NOX as a mediator in HSCs.33,35 Several experiments demonstrate that Ang II is able to induce ROS production in human activated HSCs. Ang II induces a marked increase in ROS formation in dichlorodihydroflorescein diacetate (DCFDA)-loaded activated human HSCs. Ang II-induced ROS formation in HSCs is inhibited when HSCs are treated with either a NOX inhibitor diphenylene iodonium (DPI)36 or with the AT1R antagonist, losartan.28 Furthermore, Ang II induces the phosphorylation of p47phox through AT1R in activated HSCs. In particular, Ang II leads to an increase of intracellular ROS in HSCs isolated from wild type (WT) mice, but not in p47phox-/- HSCs. Also, Ang II induces DNA synthesis and cell migration in WT HSCs, as well as phosphorylation of ERK and c-Jun, while these effects are blunted in p47phox-/- HSCs.33 Taken together, Ang II directly acts on HSCs through the activation of the NOX complex and the subsequent production of ROS. In addition, NOX is a crucial mediator of the proliferative action of PDGF and profibrogenic action of leptin.37,38 Leptin-induced NOX acts downstream of JAK activation but is independent of STAT3.38 These reports in conjunction with previous studies on Ang II, place NOX in the center of the fibrogenic signaling response in HSCs and demonstrate its potential pharmacological target for antifibrotic therapies.
The molecular mechanism of NOX activation by Ang II is still incompletely understood. In vascular smooth muscle cell, Ang II-stimulated ROS generation is biphasic.39 The fast response is dependent on protein kinase C (PKC) which phosphorylates p47phox then phosphorylated p47phox translocates to membrane where it activates NOX catalytic subunit NOX1 and/or NOX2. The slow response requires Rac activation by upstream c-Src, EGF receptor transactivation, phosphatidylinositol-3-kinase activation.39 Ang II may stimulate PKC-dependent NOX activity through three different phospholipases (PL) such as PLC, PLD, and PLA2.14 The prolonged production of NOX-mediated ROS requires upregulation of NOX catalytic subunits and components.40
Both phagocytic and non-phagocytic NADPH oxidases in hepatic stellate cells mediate hepatic firosis
In the liver, NOX is functionally expressed both in the phagocytic form and in the non-phagocytic form.41 Through analysis of mRNA expression of NOX components in isolated liver cell tractions from wild type C57BL/6 mice, we demonstrated that phagocytic NOX components such as NOX2, p40phox, p47phox, and p67phox are mainly expressed in Kupffer cells, whereas nonphagocytic NOX components including NOX1, NOXO1, and NOXA1 are expressed in HSCs and endothelial cells.42
Although NOX plays an important role in HSCs activation, the exact structure of this complex is still unknown. All components and functionalities of the phagocytic NOX are already well established, but the respective interactions between the non-phagocytic NOX components are still under investigation. p47phox is well established component as playing a central role in the activity of NOX in HSC.33 Another integral component required for the activation of NOX is Rac1. Rac1 is a member of the Rho family of small GTPase proteins that regulates cell proliferation and dynamic reorganization of the actin cytoskeleton.43
We reported that both NOX1 and NOX2 proteins are upregulated in the fibrotic liver. In particular, non-phagocytic NOX components as well as phagocytic NOX are upregulated in activated HSCs compared to quiescent HSCs.42 Wild-type HSCs express the mRNAs for both phagocytic NOX2 and non-phagocytic NOX including NOX1 and NOX4. HSCs also express other NOX components including p47phox, p67phox, p40phox, NOXO1, NOXA1, and Rac1. Both phagocytic and non-phagocytic NOX catalytic subunits including NOX1, NOX2, and NOX4 are upregulated in activated HSCs compared to quiescent HSCs. Indeed, the mRNA for the cytoplasmic factor p47phox and the cell membrane proteins NOX2 and NOX1 are detected at very low levels in quiescent HSCs by real time RT-PCR, while they are highly expressed following HSC activation in culture and in cells freshly isolated from patients with liver firosis.33,42 These data indicate that after HSCs are activated in vivo, there is activation of both phagocytic and non-phagocytic NOX components.
Regarding the contributory role of phagocytic NOX2 and non-phagocytic NOX1 in hepatic fibrosis, we observed that mice deficient for either NOX1 or NOX2 displayed significantly less hepatic fibrosis compared to WT mice after CCl4 injections or BDL.42 Through experiments of hepatic fibrosis induction in NOX1 or NOX2 bone marrow chimeric mice, we demonstrated that both NOX1 and NOX2 in endogenous liver cells including HSCs mediate hepatic fibrosis.42 Recently other investigator also reported that NOX1 mediates hepatic fibrosis by mechanism that NOX1-derived ROS oxidize and inactivate phosphatase and tensin homologue (PTEN) resulting in protein kinase B (Akt)/forkhead box O(FOXO) 4 activation and promoting HSC proliferation.44 Thus, non-phagocytic NOX as well as phagocytic NOX represents a fundamental mediator in experimental liver fibrosis.
In addition to receptor-mediated agonists such as Ang II, NOX may be activated in HSCs by the engulfment of apoptotic bodies from dying hepatocytes leading to the up-regulation of collagen ╬▒1(I) in HSCs.45 The engulfment of apoptotic bodies not only acts on macrophages stimulating production of TGF╬▓1,46 but also on HSCs promoting their activation. In support of this model, apoptotic bodies activate NOX in HSCs, thereby up-regulating HSCs activation and firosis.47 Specifically, apoptotic bodies in HSCs increase ROS production in an NOX-dependent manner. The increased production of ROS and collagen in response to apoptotic bodies is blocked by DPI, a NOX inhibitor. Jiang et al. reported that phagocytosis of apoptotic hepatocytes directly induce HSC activation and collagen production via NOX2.48 It can be presumed that NOX2 expression in HSCs may reflect the phagocytic function of HSCs.47 These results are again consistent with an important role of NOX in HSCs activation. The profibrogenic role of NOX-mediated ROS in HSCs is schematically summarized in Figure 2.
CONCLUSION
ROS has diverse effects with respect to different kinds, concentration, and cell types. Although higher concentration of ROS can be cytotoxic, lower concentration of ROS serves as second messengers during cellular responses to a variety of physiologic stimuli. Emerging evidences suggest that non-phagocytic NOX as well as phagocytic NOX in hepatic stellate cells mediate hepatic fibrosis. Because NOX2 activity is important in host defense, targeting specific components of non-phagocytic NOX could be a reasonable approach to develop antifibrotic therapies that would not suppress NOX2-mediated host defences. The molecular mechanism of redox signaling associated with various NOX homologues in hepatic fibrogenesis is still largely unknown. A better understanding of contributory role of various NOX homologues in the development of hepatic fibrosis may enable to develop novel therapeutic to prevent or treat hepatic fibrosis.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print