Meta-analysis of the correlation between the rs17401966 polymorphism in kinesin family member 1B and susceptibility to hepatitis B virus related hepatocellular carcinoma
Article information

Abstract
Background/Aims
The association between the kinesin family member 1B (KIF1B) gene polymorphism and the risk of hepatitis B virus-related hepatocellular carcinoma (HCC) has been investigated in many peer-reviewed studies. However, scholars have failed to replicate these results in validation tests. The purpose of the present study was to explore whether the KIF1B rs17401966 polymorphism was associated with susceptibility to HCC.
Methods
The results of case-controlled studies on the correlation between the KIF1B rs17401966 polymorphism and HCC susceptibility were collected using Google Scholar and the EMBASE, PubMed and CNKI databases. Based on inclusion and exclusion criteria, 5 papers with a total of 12 cohorts were included in this study.
Results
The 12 cohorts were integrated, and the results showed that the rs17401966 polymorphism reduced the risk for HCC under the allele, heterozygous, homozygous, and dominant models but not under the additive or recessive models. Moreover, the merged results showed strong heterogeneity, and the cumulative meta-analysis results were unreliable. A genetic differentiation analysis of the 12 cohorts found different degrees of genetic differentiation between the 5 cohorts in Zhang et al.’s study and the cohorts in the other studies. We further divided the 12 study cohorts into 2 subgroups based on fixation index value; however, the results of that analysis were inconsistent.
Conclusions
The results of this meta-analysis were not able to verify the association between the KIF1B rs1740199 polymorphism and HCC risk. Therefore, a well-designed, large-scale, multicenter validation study is needed to confirm the relationship.
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common malignant tumors in the world and ranks third in global cancer deaths [1]. The occurrence and development of HCC varies widely among ethnic groups, regions and individuals [2], and epidemiological investigation have shown that 60% of the more than 700,000 new HCC cases annually are caused by chronic hepatitis B virus (HBV) infection and are distributed with the epidemic characteristics of HBV [3]. In hyperendemic areas such as China and Africa, chronic HBV infection contributes to at least 80% of cases of HCC [4]. The main pathogenic factors of HCC include hepatitis C virus (HCV) infection, aflatoxin food contamination and algae toxin water contamination [5]. However, exposure to these factors rarely promotes the development of HCC, even among people who are highly exposed to the pathogenic factors of HCC, which suggests that the host’s genetic predisposition plays a crucial role in the development of HCC [6].
Assuming that certain genetic variations increase the susceptibility of chronic HBV carriers to HCC development, candidate genes have been selected a priori based on this biological likelihood. Over the past decade, candidate gene association studies have found that many genes are associated with the risk of HCC, such as TNF-α [7], IGF2 [8], SPP1 [9], DNMT3B [10]. Unfortunately, certain of these studies suffered from major methodological drawbacks because of their case–control and monocentric focus; therefore, it is difficult to validate their conclusions on HCC susceptibility. Genome-wide association study (GWAS) of human age-related macular degeneration was first reported in 2005 [11], and represented a landmark for new directions and new methods for research into complex diseases or traits. GWAS is a hypothesis-free approach that can screen disease-related sequence variations from the whole genome [12]. In 2010, Zhang et al. [13] conducted a GWAS on HBV-related HCC in the Chinese population and found a significant correlation between the single nucleotide polymorphism (SNP) locus rs17401966 in the kinesin family member 1B (KIF1B) gene of the 1p36.22 region and susceptibility to HCC, and the integrated P value of the 5 study cohorts reached 3.4×10-19. However, recent studies have yielded inconsistent or conflicting results that may have been caused by population and design differences and a small sample size. Thus, we attempted to conduct a meta-analysis of all relevant literature to provide more comprehensive and reliable associations between the rs17401966 in KIF1B gene and susceptibility to HCC with HBV infection.
MATERIAL AND METHODS
Literature search strategy and inclusion criteria
A search of case-controlled studies on the rs17401966 polymorphism of the KIF1B gene and the susceptibility to HCC published from January 2010 to April 2016 was performed in Google Scholar and in the EMBASE, PubMed and CNKI databases. The literature search was performed using the following terms: hepatitis B, chronic hepatitis B, hepatocelluar carcinoma, HCC, liver cancer, KIF1B, kinesin family member 1B, rs17401966, polymorphism and variant. KIF1B variants with an increased risk of HCC associated with the defined causes of chronic HBV infection were also identified using an online database of SNP trait associations extracted from published GWASs (http://www.genome.gov/gwastudies).
The inclusion criteria for this meta-analysis were as follows: (1) openly published case-controlled studies on the correlation between the presence of KIF1B gene polymorphism and the susceptibility to HBV-related HCC; (2) available corresponding genotype frequency data for determining differences in the populations; (3) chronic HBV carriers used as the control group; (4) exclusion of combinations with HCV and human immunodeficiency virus infection; and (5) consistent diagnosis standards for chronic HBV infection and HCC with Chinese or international standards [14,15]. The exclusion criteria were as follows: (1) family-based research; (2) unclear descriptions of the diagnosis standards of chronic HBV infection and HCC; and (3) poor quality, repeated reports, or research without detailed data.
Data collection and analysis
The literature retrieval was conducted by two independent evaluators (Mingkuan Su and Jianfeng Guo). Controversial studies were resolved through consultations or third-party evaluations. The extracted data included the author(s), publication data, sample size, genotype data, case and control group selection methods, and study group ethnicity. Both evaluators checked each other’s data to ensure the accuracy of data.
Statistical analysis
Six genetic models were adopted to analyze the correlation between the rs17401966 polymorphism and HCC susceptibility: allele (G vs. A), heterozygous (AG vs. AA), homozygous (GG vs. AA), additive (GG vs. AG), recessive (GG vs. AG+AA) and dominant (GG+AG vs. AA). The correlation was estimated by the odds ratio (OR) together with the 95% confidence interval (95% CI). The significance of the pooled OR was determined by the Z-test, and P-values of <0.05 were considered statistically significant. Cochran’s Q test was used to inspect the heterogeneity of the included studies, and an I2 quantitative determination was used to assess the degree of heterogeneity, with I2 <25% for no heterogeneity, 25%≤I2 ≤50% for mild heterogeneity, 50%≤I2 ≤75% for moderate heterogeneity and I2 >75% for strong heterogeneity. When I2 <50%, a fixed-effects model was adopted to merge the statistics; otherwise, a random-effects model was adopted. Egger’s test was used to evaluate the publication bias, and a sensitivity analysis was used to evaluate the reliability of the results. A cumulative meta-analysis was also conducted. All the statistical analyses were performed using Stata 10.0 with two-sided P-values. Population differences were estimated using Arlequin 3.5 software.
RESULTS
Characteristics of the included studies
Of the 37 retrieved references, 32 studies were excluded according to the inclusion and exclusion criteria and 5 studies were ultimately included in this meta-analysis (Fig. 1) [13,16-19]. The 12 study cohorts included in this study consisted of 8 Chinese cohorts, 2 Japanese cohorts, 1 Korean population and 1 Thai population. There were a total of 4,886 HCC cases and 5,442 controls. Subgroup 1 included 2,310 HCC cases and 1,789 controls, whereas subgroup 2 included 2,576 HCC cases and 3,653 controls. The characteristics of each study, including the number and ethnicity of the cases and controls and allele and genotype distributions, are presented in Table 1.
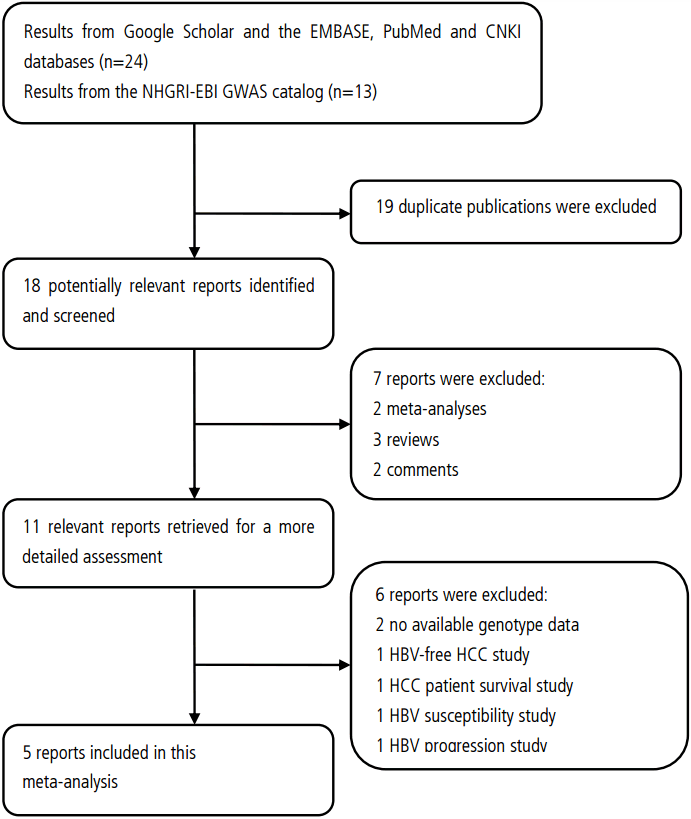
A flow diagram of the study selection process. GWAS, genome-wide association study; HBV, hepatitis B virus; HCC, hepatocellular carcinoma.
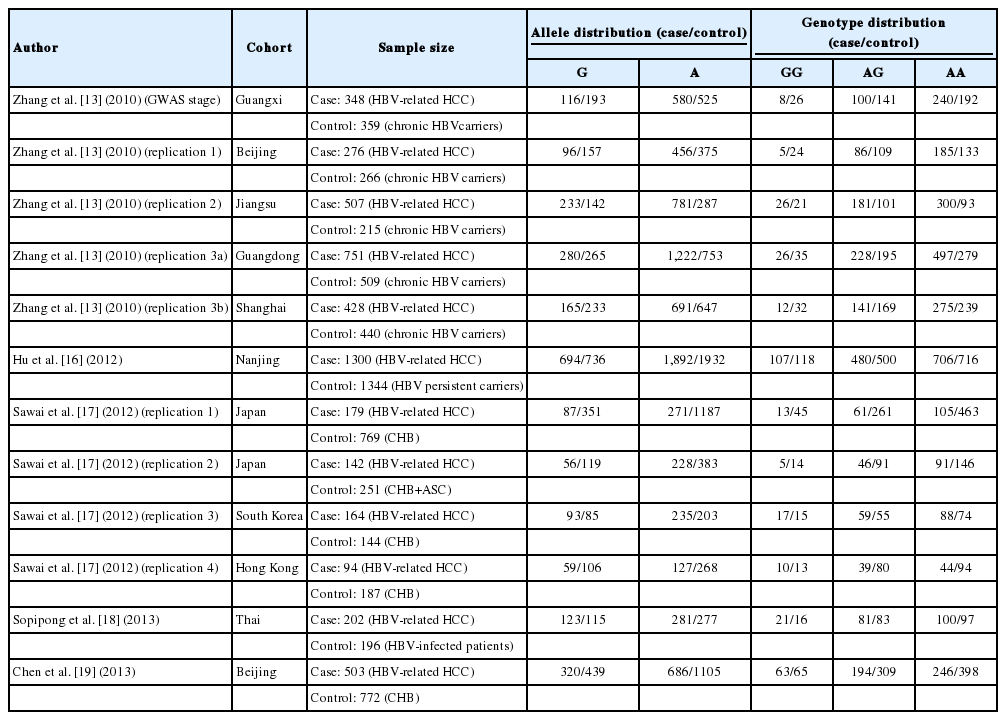
Characteristics of the included studies in this meta-analysis
Determination of population differentiation among 12 cohorts
Cohorts from different regions have different genetic backgrounds that often lead to different degrees of susceptibility to the same diseases. To avoid these hidden differences in the merged analysis, we tested the differences in the population with a fixation index (Fst) to describe the degree of genetic differentiation, with a value of Fst<0.05 between two cohorts indicating no genetic differentiation [20]. Table 2 exhibits the pairwise Fst values of the 12 cohorts. A comparison of the cohorts indicated that greater divergence occurred between the Guangxi cohort and the Hong Kong (Fst=0.064), Thailand (Fst=0.052) and Beijing 2 (Fst=0.057) cohorts; in addition, significant genetic differences were observed between the Beijing 1 cohort and the Hong Kong (Fst=0.056) and Beijing 2 (Fst=0.050) cohorts. Additional low-level genetic differences were observed among the cohorts at Fst values<0.05.

Distribution of pairwise Fstdistances between the 12 populations in HCC and control group
Association between the rs17401966 polymorphism and HCC risk among all 12 cohorts
The merged results of the 12 cohorts showed significant heterogeneity under all 6 analysis models, especially in the allele and homozygous models, which presented P-values as low as 1.67×10-10 and 3.97×10-8, respectively. Therefore, a random-effects model was used to merge the statistics. For the rs17401966 polymorphism, a decreased risk of HCC development was observed under four genetic models (G vs. A: OR=0.81, 95% CI: 0.68-0.96; AG vs. AA: OR=0.79, 95% CI: 0.68-0.91; GG vs. AA: OR=0.66, 95% CI: 0.44-0.98; GG+AG vs. AA: OR=0.77, 95% CI: 0.65-0.93), although the results of the meta-analysis were weak and presented P-values ranging from 0.002 to 0.039. Unfortunately, a significant association was not observed between the rs17401966 polymorphism and HCC risk in the additive and recessive models (GG vs. AG: OR=0.84, 95% CI: 0.64-1.12, P=0.239; GG vs. AG+AA: OR=0.72, 95% CI: 0.51-1.13, P=0.073). The results of the meta-analysis are presented in Table 3 and Figure 2.

Meta-analysis of the KIF1B rs17401966 polymorphism and HCC risk in all cohorts and subgroups by the six genetic models
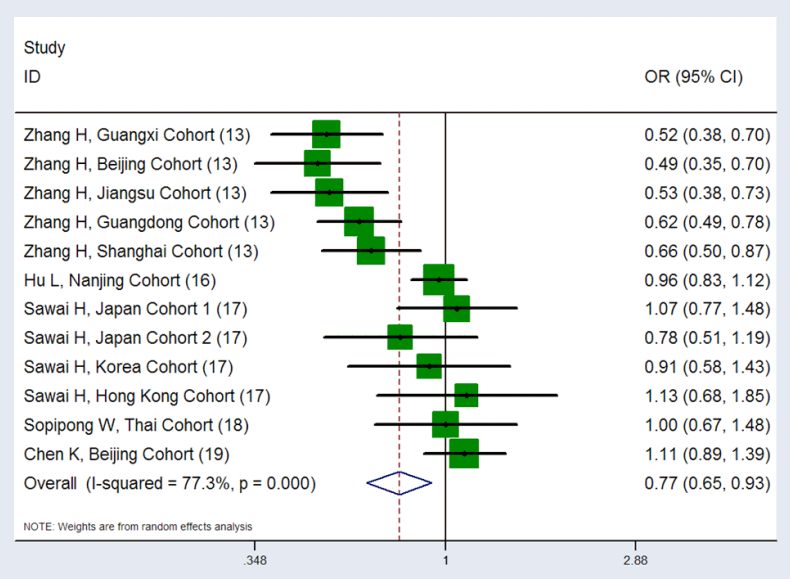
Forest plot of the association between KIF1B rs17401966 polymorphism and HCC risk, under the dominant model, merged results of the 12 included groups showed that the rs17401966 polymorphism may reduce the risk of HCC (P=0.005, OR=0.77, 95% CI: 0.65-0.93), P-value of the heterogeneity test was 1.19×10-6, I2 was 77.3%. KIF1B, kinesin family member 1B; HCC, hepatocellular carcinoma; OR, odds ratio; CI, confidence interval.
Association between rhe rs17401966 polymorphism and HCC risk in the subgroup analysis
We applied Fst statistics to determine the degree of genetic differentiation among the 12 cohorts. The I2 values of the meta-analysis of 10 cohorts (without Guangxi and Beijing 1) or 9 cohorts (without Hong Kong, Thailand, and Beijing 2) were still larger than 50%. Because there was low-level genetic differentiation between cohorts and the meta-analysis was a merged analysis of the included cohorts, a cumulative effect of the low level of genetic differentiation was observed, which produced an I2 value of >50%. Table 2 shows that the 5 cohorts of Zhang et al.’s study [13] presented different degrees of genetic differentiation compared with the other research cohorts. Therefore, we assigned Zhang et al.’s 5 study [13] cohorts to subgroup 1 and the other 7 cohorts to subgroup 2. Interestingly, we found that the rs17401966 polymorphism was significantly negatively correlated with HCC susceptibility in subgroup 1 under all 6 analysis models, especially in the allele and dominant models, which presented P-values of 6.16×10-21 and 2.89×10-17, respectively, Q-statistic values of P>0.05 and I2 values of 0.0%. In subgroup 2, the heterogeneity under the six genetic models was high and presented P-values from 0.271 to 0.983 and I2 values from 0.0% to 20.8%. Unfortunately, an association between the rs17401966 polymorphism and HCC risk was not observed under any of the genetic fixed-effects models, which presented P-values of 0.182 to 0.955.
Publication bias, sensitivity analysis and cumulative meta-analysis
Egger’s test was performed to determine whether publication bias occurred in the retrieved studies. The P-values for Egger’s test ranged from 0.151 to 0.458, which indicated that publication bias did not occur in the evaluated studies. A sen - sitivity analysis was used to evaluate the effects of individual research on the meta-analysis results, and the results for the 12 cohorts showed that the significance of the merged results did not change with the removal of any of the cohorts. Under the dominant model, the cumulative meta-analysis showed that as the included study cohort increased, the OR value showed corresponding increases (Fig. 3), and a similar trend was observed under the other 5 analysis models.
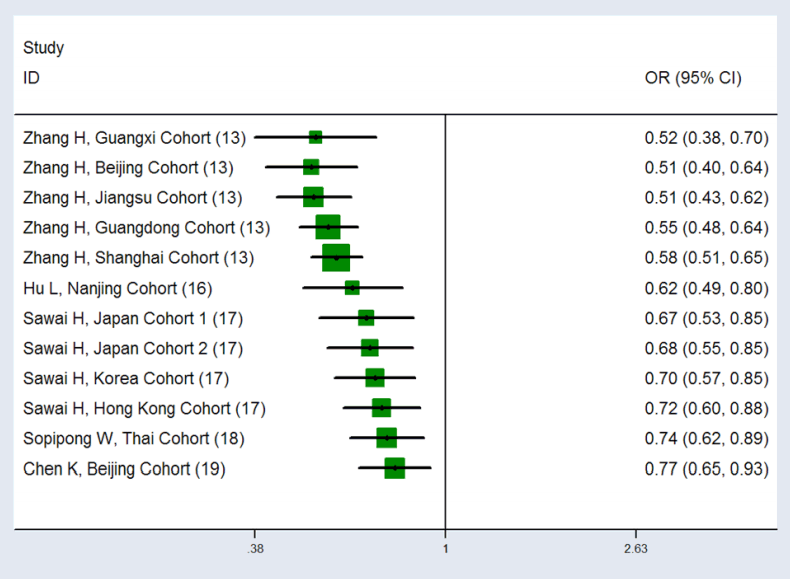
The cumulative meta-analysis for the association between KIF1B variant and HCC risk. The dominant model showed OR was increased corresponding to an increase in the included research population, suggesting unstable cumulative meta-analysis results. KIF1B, kinesin family member 1B; HCC, hepatocellular carcinoma; OR, odds ratio; CI, confidence interval.
DISCUSSION
In 2010, Zhang et al. [13] conducted a GWAS with samples from the primary screening stage from 355 chronic HBV carriers with HCC and 360 chronic HBV carriers without HCC from Guangxi, and they validated their results with four additional independent case-controlled populations recruited from Guangdong, Shanghai, Jiangsu and Beijing. The results showed that the SNP locus rs17401966 in the 1p36.22 region was verified in the four populations and had a merged P-value of 3.4×10-19. The 1p36.22 region was approximately 244 kb long, including KIF1B, phosphogluconate dehydrogenase (PGD) and the 3’ terminal end of the ubiquitination factor E4B (UBE4B) genes. 1p36 is an important loss of heterozygosity (LOH). LOH has been frequently observed in tumor originated from the blood cells, nerve cells and epithelial cells [21]. In addition, Li et al. [22] found in the study of HCC patients in the group of southern China that there is high frequency of LOH in 1p36 of cancer tissues, suggesting this area is correlated with the occurrence of HCC. Thus, the newly identified UBE4B-KIF1B-PGD locus is a biologically plausible candidate for HCC susceptibility. Zhang et al. [13] also performed an immunohistochemical analysis to determine the protein expression of the KIF1B, PGD and UBE4B genes in the HCC tissues and the corresponding paracarcinoma tissues, and although the results did not indicate abnormalities in the expression of UBE4B, a significant reduction in the expression of KIF1B and PGD was observed in the cancerous tissue. A qRT-PCR analysis then showed that the expression of KIF1Bβ was correlated with the genotype of the associated SNP locus rs17401966 and indicated that the high expression of this gene was also significantly related to the protective allele G of rs17401966, which suggests that KIF1Bβ may represent a HCC-suppressor gene. KIF1B is a member of the kinesin family and an important molecule in intracellular vesicles and organelles transport [23], in combination with the absence of the adjacent gene on 1p36 (e.g., p73 and CHD5), the down-regulation of KIF1Bβ may lead to the occurrence of tumors [24,25].
In 2012, Li et al. [26] conducted a GWAS that included two independent Han cohorts at the genome-wide discovery stage, and they covered 480 HBV-positive HCC patients and 484 chronic HBV carriers from central China and 1,058 cases and 981 controls from southern China. These authors assessed the previously reported HCC-susceptible SNP rs17401966; however, the merged results for the two groups showed that the rs17401966 G allele did not reduce the risk of HCC (OR=0.90; 95% CI: 0.80-1.02). In 2013, Jiang et al. [27] conducted another GWAS with 1161 cases and 1,353 controls from Qidong at the genome-wide discovery stage, although the findings were not confirmed (OR=0.98; 95% CI: 0.87-1.11). Subsequent studies by other scholars also failed to achieve consistent results [16-19]. Because the role of the same gene can vary among different populations, among the same group and among with the same tumor, a single study may not provide enough samples for a correlation analysis or sufficient statistical validity to identify minor genes, the meta-analysis approach expands the sample size to avoid the flaws of insufficient statistical validity. In this meta-analysis, we included a total of 12 cohorts from 5 studies, and the results showed that the rs17401966 polymorphism might reduce the risk of HCC under the allele, heterozygous, homozygous, and dominant models but not under the additive and recessive models. The inconsistent results might have been caused by the lack of advantage observed in the homozygous rs17401966 GG with regard to a reduction in HCC susceptibility compared with that observed for the heterozygous rs17401966 AG. However, strong heterogeneity was observed in the merged analysis of the 12 cohorts, which might reduce the reliability of the results. The Fst statistics indicated that different degrees of differentiation occurred between the 5 cohorts of Zhang et al.’s study [13] and the other cohorts. To reduce the effect of Zhang et al.’s study [13] cohorts on the meta-analysis, we assigned them to subgroup 1, and the remaining seven cohorts were assigned to subgroup 2. Inconsistent meta-analysis results were observed between subgroup 1 and subgroup 2, which may been caused by the following reasons: (1) the 7 cohorts of subgroup 2 were from different studies that presented differences in their case selection, such as the genetic background of the chronic HBV carriers and chronic hepatitis B; (2) strong differentiation was observed between the Beijing population included in Zhang et al.’s study [13] and Chen et al.’s study [19], which might have been caused by the accelerated population migration that occurred as China’s society and economy developed or by sampling error; and (3) the HCC sampled in different areas was caused by different genes, and the KIF1B gene mutations may not have played a dominant role in the development of HCC in certain areas.
Two meta-analyses [28,29] on the association between the KIF1B rs17401966 polymorphism and HCC risk concluded that the rs17401966 G polymorphism could reduce the susceptibility to HCC; however, the source of the heterogeneous results was not evaluated in these studies. We investigated the source of heterogeneity with an Fst analysis and conducted appropriate groupings. In our meta-analysis, although publication bias was not observed and the sensitivity analysis results did not show significant changes in the merged results when any one cohort was excluded, the results were still unreliable because the cumulative meta-analysis showed that the OR value increased as the number of research cohorts increased. It is important to note that the sample size at the primary screening stage in the GWASs of Li et al. [26] and Jiang et al. [27], as well as in the GWASs of Hu et al. [16] and Chen et al. [19], were all larger than that of Zhang et al. [13]. In addition, the sample size of subgroup 2 was greater than that of subgroup 1, and the results were inconsistent. Therefore, we propose that further studies are required to verify the correlation between the KIF1B rs17401966 polymorphism and HCC risk.
Certain limitations of this meta-analysis should be acknowledged. First, for the HBV in China, priority is assigned to genotypes B and C, although most studies do not analyze these genotypes separately. Because the information was incomplete, the above factors were not considered. Second, the studies of Li et al. [26] and Jiang et al. [27] lacked genotype data; therefore, we did not include them in the meta-analysis and the results might be biased.
In conclusion, we introduced Fst statistics to evaluate the source of heterogeneity for a binary variable meta-analysis and conducted a merged analysis on 12 cohorts, and our results indicated that the rs17401966 polymorphism can reduce the susceptibility to HCC. However, the meta-analysis results were weak and the cumulative meta-analysis results were unreliable. In addition, the results of the two subgroups were inconsistent. A well-designed multicenter validation study with a larger sample size is needed to verify this correlation.
Notes
Funding Support
The study was supported by the grants from Fujian Natural Science Fund (2016J01596) and Ningde Science and Technology Plan Project (20150013).
Conflicts of Interest: The authors have no conflicts to disclose.
Abbreviations
Fst
fixation index
GWAS
genome-wide association study
HBV
hepatitis B virus
HCC
hepatocellular carcinoma
KIF1B
kinesin family member 1B
SNP
single nucleotide polymorphism