INTRODUCTION
Alagille syndrome (AGS) is an autosomal-dominant disorder with varying degrees of abnormalities in the liver, heart, eyes, face, bone, and to a lesser degree the kidney, vasculature, and pancreas [1-4]. The majority of patients with AGS become symptomatic in infancy or early childhood [5]. A classical diagnosis of AGS has been based on the finding of intrahepatic bile duct paucity on liver biopsy associated with three to five major features: chronic cholestasis, cardiac disease, skeletal abnormalities, ocular abnormalities, and characteristic facial features [1,3].
The traditional clinical criteria have been challenged with the emergence of molecular screening, in fact AGS is known to be caused by mutations in one of two genes, namely, JAG1 or NOTCH2 [4,6,7]. These genes are part of the Notch signaling pathway, which is involved in cell fate determination. JAG1 mutations have been identified in 70ŌĆō94% of individuals with clinically diagnosed AGS [8-10]; while several NOTCH2 mutations have been reported to date [7,8,11]. Transmission follows in an autosomal dominant pattern, with a high degree of penetrance but variable expressivity [5,12]. In an atypical or mild case, molecular confirmation of the diagnosis of AGS is valuable not only for genetic counseling purposes but also for diagnosis itself [9].
In this report, we describe the case of AGS diagnosed in oneŌĆÖs adulthood presenting chronic cholestatic feature.
CASE REPORT
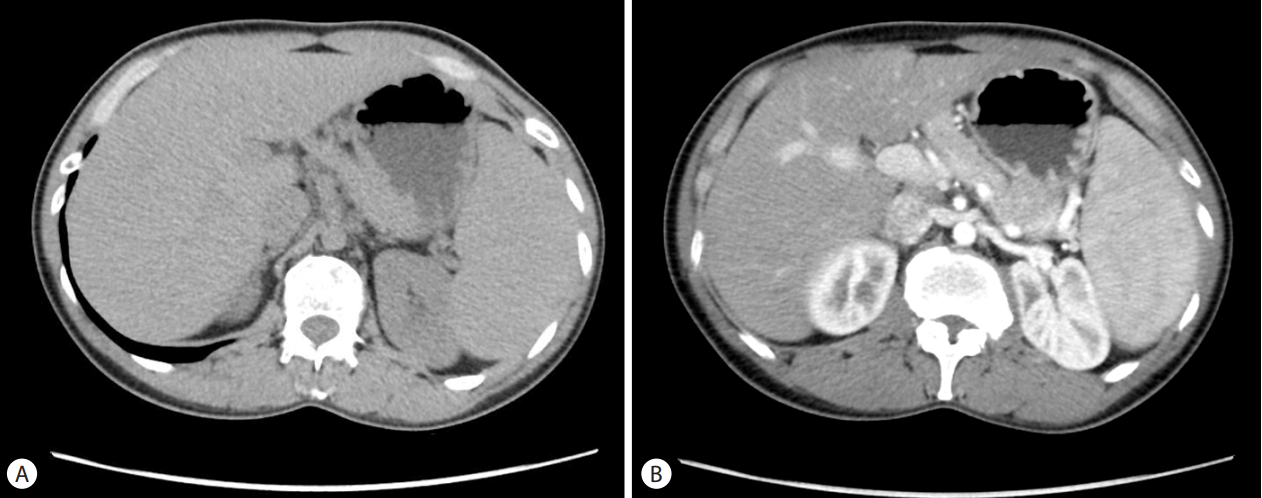
A 31-year-old male patient was referred to our center with abnormal liver enzyme level at army physical readiness test 6 months ago. Before coming to our center, he had several tests including liver biopsy, which were insufficient to determine the cause of gamma-glutamyl transpeptidase (GGT) and alkaline phosphatase (ALP) elevation. He had an unremarkable medical history except receiving surgery due to recurrent left pneumothorax in his teen age. The first laboratory findings at our hospital in February 2013 were as follows: total bilirubin 1.7 mg/dL; aspartate aminotransferase (AST) 78 IU/L; alanine aminotransferase (ALT) 39 IU/L; ALP 308 IU/L; GGT 542 IU/L; international normalized ratio (INR) 0.88. Immunoglobulin G (IgG) level was within reference range (700-1600 mg/dL). Serum copper as screening test for WilsonŌĆÖs disease was normal as 129 ╬╝g/dL (Reference range: 55-150 ╬╝g/dL). Also serum anti-mitochondrial antibodies (AMA) which may be indicative of primary biliary cirrhosis was negative. All the serological markers for viral hepatitis were not remarkable. Liver fibroscan revealed borderline fibrosis with 8.6 kPa and minimal steatosis with controlled attenuation parameter (CAP) level of 227 dB/m. Computed tomography (CT) showed findings consistent with chronic liver disease, without focal nodule or abnormal vascularity (Fig. 1). The result of the first liver biopsy at other tertiary hospital was nonspecific as follows: chronic hepatitis with mild lobular activity, mild periportal activity and septal fibrosis. Our pathologist read that biopsy slide similarly but also noted following findings: rare bile duct in portal tracts.
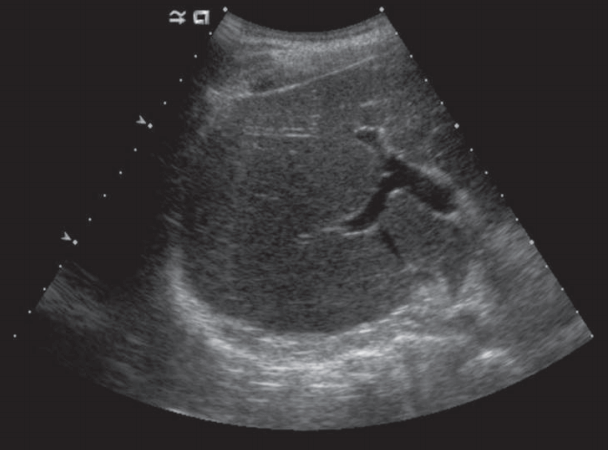
After three months, liver enzymes showed nonspecific changes: AST slightly increased to 82 IU/L; GGT decreased to 250 IU/L; total bilirubin changed from 1.7 to 2.3 mg/dL. Anti-smooth muscle antibody test showed positive result but at low titer (1:20). Three months later, total bilirubin showed a slight decline but GGT increased to 357 IU/L. Abdominal ultrasonography as follow-up test showed coarse liver parenchymal echogenicity, a few tiny gall bladder polyps, splenomegaly, and renal parenchymal disease but no bile duct dilatation (Fig. 2). Then the patient began to take high-dose ursodeoxycholic acid (300 mg three times a day) and kept routine check-up every 3 months.
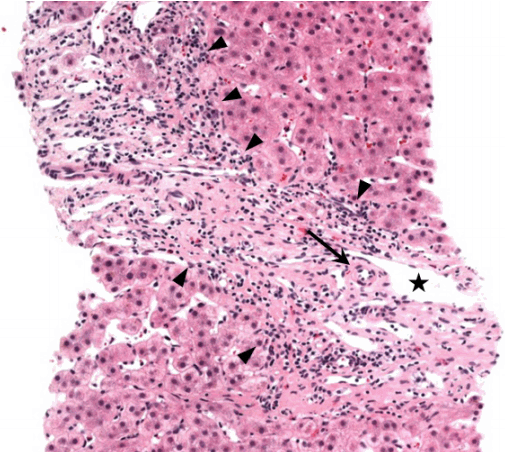
During 10-month follow-up period, ALT increased to 565 IU/L and GGT level remained high. IgG increased steadily to 2011 mg/dL. Serum ALP isoenzyme electrophoresis report disclosed that ALP was mainly derived from liver (Liver 94.64%, Bone 5.36%). So we decided to repeat liver biopsy. In August 2014, liver biopsy revealed chronic hepatitis with undetermined etiology. The result contains the followings: mild to moderate intralobular and piecemeal necrosis; moderate portal inflammation; bridging fibrosis; fatty change below 5%; predominantly lymphoplasmacytic infiltration; rare bile ducts in portal tracts ŌĆō complete portal tracts numbered ten (Fig. 3). It was almost the same as the result of previous liver biopsy.
After three monthsŌĆÖ therapy with the same oral medication, liver enzyme profile didnŌĆÖt change significantly. Then we thought about rare genetic diseases like vanishing bile duct syndrome. We made a consultation to our laboratory medicine specialist for genetic counseling but the patient refused to undergo whole exome sequencing test because of economic burden. Then 11 months later, in November 2015, there is still no big difference in follow-up laboratory findings. Actually the patient had some kind of unusual facial findings such as prominent forehead, deep-set eyes with mild hypertelorism, a straight nose, a pointed chin and large ears. We hit upon AGS so consulted our pediatrician and performed JAG1 gene mutation analysis. By direct sequencing of genomic DNA (total 26 exons and adjacent introns within chromosome 20p12) isolated from peripheral blood leukocytes, JAG1 gene mutation was probably detected.: G, the first base sequence of intervening sequence 19, was replaced by T (NM_000214.2:c.2372+1G>T). This mutation hasnŌĆÖt been reported yet. But regarding that the mutation is located in consensus splice-donor site, it has certain potential for leading to a genetic disease by affecting the splicing process.
Spine radiographs of the patient showed mild hyperostosis of whole spine and also subchondral erosion and sclerosis of both sacroiliac joints. The patient didnŌĆÖt have any cardiac abnormalities in screening echocardiography. The patientŌĆÖs father underwent surgery for aortic valve disease in December 2015, but had normal liver function. The other family members ŌĆō mother and younger sister ŌĆō had no specific medical history. These three family members didnŌĆÖt look similar to the patient.
A diagnosis of AGS was made on the combination of the presence of genetic mutation, the result of liver biopsy, characteristic facial features, cholestasis and skeletal abnormalities. We keep on ursodeoxycholic acid therapy and routine check-up with blood test and liver imaging studies.
DISCUSSION
As described above, AGS is a genetic condition that is characterized by chronic cholestasis due to a paucity of bile ducts and multi-organ involvement of varying severity [1,5,12]. Neonatal cholestasis is a main feature and it can progress to cirrhosis [3,13,14]. Because the manifestations are predominantly pediatric, a diagnosis of AGS is usually made in early childhood [1].
To find the cause of cholestasis is often difficult in clinical practice, especially after ruling out common diseases. In the case presented here, ALT and GGT were dominantly elevated and hyperbilirubinemia was minimal in an adult patient without definite history of hepatobiliary disease. Comprehensive laboratory tests targeting relatively common cholestatic diseases in adult patient were done but the results were negative. A diagnosis of AMA negative primary biliary cirrhosis (PBC) couldnŌĆÖt be made, because the pathology revealed ductopenia rather than typical bile duct destruction as shown in PBC and the patient had no pruritus, jaundice, dry eye or elevated serum level of immunoglobulin M [15]. Idiopathic adulthood ductopenia (IAD) is another cholestatic liver disease with biopsy-proven ductopenia. But it isnŌĆÖt associated with multi-organ involvement including distinguishing facial features. As the term ŌĆśidiopathicŌĆÖ implies, the etiology of IAD is unknown and a diagnosis of the disease requires exclusion of other conditions with chronic cholestasis [16]. Most doctors dealing with adult patients are not used to rare syndromic pediatric diseases. ThatŌĆÖs why the diagnosis was difficult in this case.
Genetic mutation test was very helpful in confirming the diagnosis of this case. Classically, a diagnosis of AGS was based on the presence of intrahepatic bile duct paucity on liver biopsy with at least three out of five major features: chronic cholestasis, cardiac disease, skeletal abnormalities, ocular abnormalities, or dysmorphic facial features [1]. Recently, even a liver biopsy is unnecessary [8,10]. Because the mutations in JAG1 gene or NOTCH2 play a major role in pathogenesis of the syndrome, whether these mutations exist or not can be a significant feature with clinical symptoms aside. The JAG1 gene is located within band 20p12 and consists of 26 exons that cover 38,000 kb of genomic DNA. The frequency of identifiable genetic mutations in clinically diagnosed AGS patients is high, with JAGGED1 (JAG1) mutations identified in 94% and NOTCH2 mutations in 2% of patients [4,6,7]. Though AGS is inherited in an autosomal dominant manner, a substantial portion of AGS is sporadic. In this case, the patientŌĆÖs family members have no definite features associated with AGS. According to previous reports, approximately 30%-50% of affected individuals have an inherited pathogenic variant and about 50%-70% have a de novo pathogenic variant [10,17,18].
From the medical record, we found that the patient had visited our otology department by himself because of chronic otitis and cholesteatoma in December 2015. The correlation of AGS and chronic otitis has not been established but Quiros-TejeiraŌĆÖs group reported the high incidence of chronic otitis media in AGS patients [19]. We think chronic otitis can be a manifestation of AGS.
To our knowledge, this is the first report of AGS diagnosed in oneŌĆÖs adulthood. Although the cholestasis was rather mild, the diagnosis was meaningful in terms of predicting prognosis and knowing transmission pattern. Given that the AGS phenotype shows variable expression ranging from mild cholestasis to acute liver failure on cirrhosis, we should think of it as a differential diagnosis for cholestasis with indistinct cause.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print