Adefovir-induced Fanconi syndrome associated with osteomalacia
Article information

Abstract
Fanconi syndrome is a dysfunction of the proximal renal tubules that results in impaired reabsorption and increased urinary loss of phosphate and other solutes. The pathophysiology of drug-induced Fanconi syndrome is unclear. Here we report the case of a 36-year-old woman who presented with pain in multiple bones and proteinuria. She had a 7-year history of taking adefovir at 10 mg/day for chronic hepatitis B. Three years previously she had received surgery for a nontraumatic right femur neck fracture, after which she continued to complain of pain in multiple bones, and proteinuria, glycosuria, and phosphaturia were noted. The findings of a light-microscope examination of a renal biopsy sample were normal, but mitochondrial damage of the proximal tubules was evident in electron microscopy. Western blot analysis revealed that the level of serum fibroblast growth factor 23 (FGF23) was lower than in normal controls. After 2 months of treatment, hypophosphatemia and proximal tubular dysfunction were reversed, and serum FGF23 had normalized. This case suggests that direct mitochondrial damage in proximal tubules can cause drug-induced Fanconi syndrome associated with osteomalacia.
INTRODUCTION
Adefovir has been widely used for the treatment of hepatitis B and human immunodeficiency virus (HIV) infections [1,2]. Several side effects associated with adefovir are reported, including acute renal failure and Fanconi syndrome (FS) [3,4]. FS results from dysfunction of reabsorption in the proximal tubules of the kidney. This impairment causes increased excretion of solutes, such as amino acids, glucose, uric acid, bicarbonate, and phosphate [5]. Phosphaturia, in particular, leads to osteomalacia which results in symptoms such as muscle weakness, fatigue, bone pain, and fractures [6].
The mechanism of drug-induced FS is unclear. Fibroblast growth factor 23 (FGF23) is an important hormone involved in the regulation of serum phosphate levels. Whether FGF23 contributes to hypophosphatemia due to drug-induced FS is unknown. An association between FS and FGF23 has been reported, but the findings have been inconsistent [7–11]. Pathogenesis of drug-induced FS may involve mitochondrial damage in the proximal tubules [12].
We evaluated a woman with chronic hepatitis B receiving adefovir who presented with proteinuria, hypophosphatemia, progressive bone pain throughout the body, and bone fractures. Eventually, she was diagnosed with FS associated with adefovir. FGF23 levels and mitochondria in proximal tubules were evaluated. Here, we report the FGF23 levels and results of renal biopsy in the case of adefovir-induced hypophosphatemic osteomalacia. Our data suggest that mitochondrial damage in the proximal tubules is critical in the pathogenesis of drug-induced FS.
CASE REPORT
A 36-year-old woman visited the nephrology department of Soonchunhyang University Cheonan Hospital with progressive multiple bone pain and proteinuria. She was not independently mobile and was using a wheelchair. She had a 9- and 7-year history of 100 mg/day lamivudine and 10 mg/day adefovir, respectively, for chronic hepatitis B. Three years previously, she received surgery for a non-traumatic right femur neck fracture. A 99mTcbone scan was performed due to muscle weakness and multiple bone pain. Multiple hot bone uptakes were noted in the right clavicle, both ribs, and pelvis (Fig. 1A). Dual-energy X-ray absorptiometry revealed decreased lumbar spine bone mineral density of 0.786 g/cm2 (T-score, −2.8; Z-score, −2.5).

99mTc-bone scan and serum FGF23 level in patients. (A, B) 99mTc-bone scan showing multiple hot spots of bone uptake in the right clavicle, both ribs, and pelvis before (A) and after (B) 8 months of treatment. (C) Validation of FGF23 antibody levels using immunoblotting. Purified human FGF23 (hFGF23) was detected by Western blotting. (D) Serum FGF23 levels of the patient, the patient’s sister (Patient’s Rel.), and an unrelated normal subject (Normal Ctr.) at admission. (E) Serum FGF23 levels at administration (Admin.) and after treatment (8 and 10 months). Rel., relative; Ctr., control.
On admission, multiple bone pain with tenderness was noted on physical examination, and vital signs were unremarkable. The patient’s weight and body mass index were 47.4 kg and 18.9 kg/m2, respectively. She denied a history of hypertension and diabetes, and the use of any other medications. The results of serology markers for quantitative surface antigen of hepatitis B virus (HBsAg) was 6,481 cutoff index (COI) with positive hepatitis E antigen, negative hepatitis E antibody, and undetectable hepatitis virus B DNA (HBV DNA). Laboratory analyses revealed white blood cell count, 4,540/μL; hemoglobin, 13.8 g/dL; hematocrit, 41.2%; platelet count, 289,000/μL; total bilirubin, 0.3 mg/dL; aspartate aminotransferase, 17 IU/L; alanine aminotransferase, 12 IU/L; lactate dehydrogenase, 150 IU/L; high-sensitivity C-reactive protein, 1.22 mg/L; prothrombin time, 12.1 seconds; activated partial thromboplastin time, 37.8 seconds; serum osmolality, 287 mOsm/kg; and osmolar gap, 2 mOsm/kg. Serum creatinine level was within normal limits and marked hypophosphatemia and hypouricemia were noted (Table 1). The elevated alkaline phosphatase implied the presence of metabolic bone disease, but intact parathyroid hormone and vitamin D levels were noted within normal limits. Arterial-blood gas analysis values were as follows: pH, 7.386; PaCO2, 35.7 mmHg; PaO2 110.4 mmHg; HCO3-, 20.9 mEq/L; base excess, -3.5 mEq/L; O2 saturation, 98.0%; and anion gap, 12.1 mEq/L. Urinalysis with microscopic examination showed pH, 7.0; specific gravity of 1.012; trace protein; two positives of glucose; 5 to 9 red blood cells per high-power field; and 1 to 4 white blood cells per high-power field. Twenty four-hour urine analysis revealed generalized amino-aciduria and impaired reabsorption of other solutes, indicating proximal tubule dysfunction (Table 1). Blood aluminum levels and 24-hour urine aluminum levels were 2.38 μg/L and 2.2 μg/day, respectively. Antinuclear antibody, antibody to double-stranded DNA, and anti-neutrophil cytoplasmic antibody were all negative. Hormone levels measured at 8:00 AM revealed the following: adrenocorticotropic hormone, 9.71 pg/mL; cortisol, 8.13 μg/dL; luteinizing hormone, 3.39 mIU/mL; follicle stimulating hormone, 5.53 mIU/mL; and estradiol 53.23 pg/mL. There was no evidence of dysproteinemia. Renal biopsy confirmed normal renal pathology in light microscopy, and immunofluorescence stains were negative. In electron microscopy, the glomerulus appeared normal but there was mitochondrial damage in proximal tubules; structures in distal tubules were near normal (Fig. 2). Finally, we made a diagnosis of FS. To measure serum FGF23 levels, immunoblotting using a standard curve with FGF23 validation of purified human FGF23 antibody (Fig. 1C) was performed [13]. Serum FGF23 levels were lower than those of normal controls (Fig. 1D).

Biochemical data at admission and 8 months after adefovir withdrawal
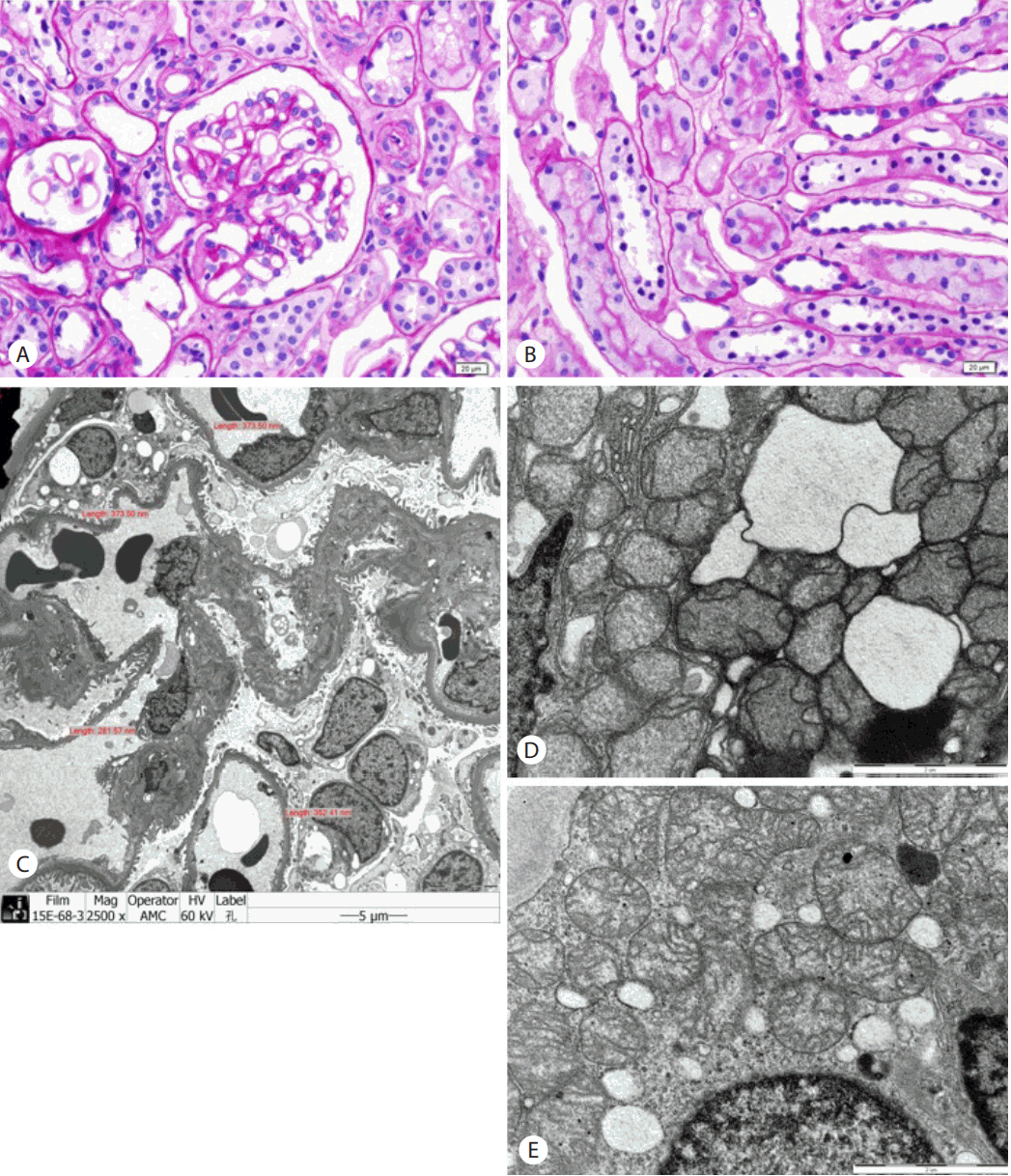
Renal pathology of the patient. The glomerulus (A) and tubules (B) were normal in light microscopy (Periodic acid-Schiff stain, the bar represents 20 μm). (C) There were nonspecific findings in conventional electron microscopy performed to detect the presence of electron-dense deposits or microstructures in patients with suspicion of glomerulonephritis (the bar represents 5 μm). (D) Severe mitochondrial damage of proximal tubules was revealed in focused electron microscopy. (E) The structures of mitochondria in distal tubules had a near-normal appearance (in picture D and E, the bar represent 2 μm).
Adefovir and lamivudine were replaced with 1 mg/day of entecavir, and phosphate (2,000 mg/day of K-phos original tablet containing 114 mg of phosphorus and 144 mg of potassium in a 500 mg tablet) and 0.25 μg/day of calcitriol were added. After 2 months of treatment, the level of serum phosphate was markedly increased (3.4 mg/dL). The patient’s bone pain was markedly improved and she was able to perform normal daily activities and could walk again without a wheelchair. After 8 months of treatment, the ratio of the renal tubular maximum reabsorption rate of phosphate to the glomerular filtration rate (TmPi/GFR) was increased to 2.31 mg/dL, and other laboratory markers indicating proximal tubule dysfunction were normalized (Table 1). Multiple hot uptakes apparent in the first 99mTc-bone scan had disappeared (Fig. 1B) and serum FGF23 level had normalized (Fig. 1E). The quantitative HBsAg was 5,292 COI without hepatitis B DNA detection, and the status of hepatitis E antigen and antibody was not changed after switch to entecavir.
DISCUSSION
We report a case with mitochondrial damage in the proximal tubules and decrease of the FGF23 levels in a patient with adefovir-induced hypophosphatemic osteomalacia and chronic hepatitis B infection. FS can present as hypophosphatemic osteomalacia, which is attributed to the impaired reabsorbing phosphate in the proximal renal tubule [6]. Nephrotoxicity associated with high dose of adefovir, 120 mg/day, used in patients with HIV is well known [1]. Adefovir has an adverse event profile similar to that of a placebo when used at a dose of 10 mg/day in patients with chronic hepatitis B infections [2]. However, nephrotoxicity including FS and hypophosphatemic osteomalacia has been described in several reports in patients taking a low dose of 10 mg/day [3,4].
Presently, the patient’s symptoms were nonspecific. Symptoms including myalgia, general weakness, or multiple fracture without an explainable cause have been described [3,4]. In our opinion, suspicion of FS is most important to make a diagnosis, and urinalysis is the most valuable screening test for FS. FS should be considered in non-diabetic patients when chronic glycosuria is evident and when a tubular range of proteinuria exists or when the levels of phosphorus and uric acid are lower than normal range or in the lower normal margin. The temporal relationship between exposure to medications with known proximal tubule toxicity and the development of tubulopathy [12] suggests the possibility of FS. In such patients, diagnosis as FS is aided by evaluation for excretion of phosphorus or uric acid, such as fractional excretion of uric acid, fractional excretion of phosphate, fractional tubular reabsorption of phosphate, and TmPi/GFR [12].
FGF23 is a principle hormone regulating renal phosphate handling, and produced by osteocytes and osteoblasts in bone. In chronic kidney disease (CKD), FGF23 is increased, which enhance the excretion of phosphate and in turn, restores normophosphatemia [14]. In contrast, FGF23 levels are decreased in hypophosphatemic patients with vitamin D deficiency, FS, and Cushing’s syndrome by an ectopic ACTH-producing tumor [7]. The present study showed that serum FGF23 level was markedly decreased in this patient with severe hypophosphatemia prior to treatment and normalized after treatment. The findings from previous study are inconsistent. Whereas normal levels of FGF23 were reported in patients treated with tenofovir in one study [8], another study described elevated FGF23 in tenofovir-related hypophosphatemia [9], and with adefovir treatment [10]. Yet, a recent study by Goto S, et al. described low FGF23 levels in 2 patients with adefovir-induced FS [11]. Among 5 patients in the previous study above, 2 patients received tenofovir due to infection with HIV, and the other patients received adefovir due to chronic hepatitis B infection. The collective data suggest that FGF23 levels may not be strictly associated with either tenofovir or adefovir, or the infecting virus. We suggest that FGF23 does not influence the phosphorus level in secondary FS.
FGF23 could be affected by many contributing factors. These include renal function [15] and the amount of dietary phosphorus [16]. The most important reason for the conflicting results to date is probably the differing glomerular filtration rate (GFR), since GFR is a major determinant of FGF23 levels [15]. In previously reported cases, all the patients with secondary FS displayed mild to moderate renal impairment, in which GFR were approximately from 50 to 70 mL/min/1.73m2, referred to CKD stage between 2 to 3 [17]. In one case involving a 75-year-old woman, estimated GFR using the Modification of Diet in Renal Disease (MDRD) formula was 72.9 mL/min [10]. Thus, there is a possibility that estimated GFR using the MDRD formula could overestimate renal function, especially in more elderly woman. Conversely, our patient was young adult with normal renal function and did not have any other cause, such as hypertension, diabetes, or history of any other medications, except adefovir.
The renal biopsy revealed mitochondrial damage in the proximal tubules and no abnormalities in the glomerulus and distal tubules. After cessation of adefovir, the impaired solute reabsorption in the proximal tubules and the symptoms suggestive of osteomalacia disappeared. Thus, we suggest that mitochondrial damage in the proximal tubules is critical in drug-induced FS. In addition, these results suggest that FGF23 does not influence the occurrence of hypophosphatemia in drug-induced FS, and is independently influenced by factors including blood phosphorus level, amount of phosphorus intake, and GFR in FS.
In conclusion, adefovir induces proximal tubular mitochondrial injury and subsequent proximal tubular phosphate wasting, which results in hypophosphatemia and osteomalacia. The decreased level of FGF23 is a consequence, not a cause, of hypophosphatemia.
Abbreviations
CKD
chronic kidney disease
COI
cutoff index
FGF23
fibroblast growth factor 23
FS
Fanconi syndrome
GFR
glomerular filtration rate
HBsAg
surface antigen of hepatitis B virus
HBV DNA
hepatitis B virus DNA
HIV
human immunodeficiency virus
MDRD
Modification of Diet in Renal Disease
TmPi/GFR
ratio of the renal tubular maximum reabsorption rate of phosphate to the glomerular filtration rate
Notes
Authors’ contribution
Park S.E. and Lee E.Y. have a responsibility as the first author and the corresponding author, respectively. Kim W.I. and Cho D.H. acquired the clinical data. Kim Y.J. and Kim H.S. helped to diagnose Fanconi syndrome and to exclude other possible diseases. Kim J.H. and Cha S.K. evaluated and interpreted the level of FGF23. Park K.S. and Lee J.H. analyzed the result of light microscopy and electron microscopy. Lee S.M. adviced to lead a diagnosis of Fanconi syndrome by interpreting bone scan.
Notes
Financial support
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1A6A1A03032522, 2017R1D1A3B03027898), Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI) funded by the Ministry of Health & Welfare (HI17C-2059-010017), and Soonchunhyang University Research Fund.
Notes
Conflicts of Interest: The authors have no conflicts to disclose.