Sarcopenia: Ammonia metabolism and hepatic encephalopathy
Article information

Abstract
Sarcopenia (loss of muscle mass and/or strength) frequently complicates liver cirrhosis and adversely affects the quality of life; cirrhosis related liver decompensation and significantly decreases wait-list and post-liver transplantation survival. The main therapeutic strategies to improve or reverse sarcopenia include dietary interventions (supplemental calorie and protein intake), increased physical activity (supervised resistance and endurance exercises), hormonal therapy (testosterone), and ammonia lowering agents (L-ornithine L-aspartate, branch chain amino acids) as well as mechanistic approaches that target underlying molecular and metabolic abnormalities. Besides other factors, hyperammonemia has recently gained attention and increase sarcopenia by various mechanisms including increased expression of myostatin, increased phosphorylation of eukaryotic initiation factor 2a, cataplerosis of α ketoglutarate, mitochondrial dysfunction, increased reactive oxygen species that decrease protein synthesis and increased autophagy-mediated proteolysis. Sarcopenia contributes to frailty and increases the risk of minimal and overt hepatic encephalopathy.
SARCOPENIA CIRRHOSIS EPIDEMIOLOGY
Sarcopenia includes loss of muscle mass and/or strength leading of physical derangements and frailty [1,2]. Presence of cirrhosis is an important risk factor for the development of sarcopenia as a result of metabolic derangements caused by hepatocyte dysfunction. Sarcopenia is a major complication in cirrhosis and almost 50 to 70% of them develops sarcopenia [3]. The prevalence rates are much higher in males (50–70%) in comparison to female cirrhosis (≤20%) [4]. The severity and prevalence of sarcopenia in cirrhosis correlate with Child-Pugh’s grade [5]. Sarcopenia when added to model for end-stage liver disease (MELD) score has improved the utility for predicting survival and is particularly useful in low MELD score patients (MELD below15) [6,7].
Sarcopenia contributes to fatigue, limits exercise tolerance and may have profound impact on performance status and activities of daily living [8]. Prognostic significance of sarcopenia in decompensated cirrhosis is well reported [9-11]. Major clinical consequences include higher wait-list mortality, poor quality of life (Table 1) [12], higher morbidity and mortality after surgery in cirrhosis; and increased risk of infections [13] and hepatic encephalopathy (HE) [14]. Moreover, sarcopenia itself may contribute to liver disease progression, as a result, altered metabolism. Sarcopenia is also known to increase the length of intensive care unit and hospital stay after liver transplantation [15]. Sarcopenia is one of the complications that does not reverse post-liver transplantation and may actually even worsen due to use of steroids and calcineurin inhibitors [16]. Therefore it is critically important to identify strategies for improving muscle mass in advanced cirrhosis. Data from more recent trials have revealed that reversal of sarcopenia is associated with improved survival and lesser complications [4,17,18].

Prognostic scoring of sarcopenia in patients with liver cirrhosis in various studies
Despite this, the understanding on the pathophysiology of sarcopenia is still limited and treatment options proposed are not well explored in large randomized controlled trials. This review summarizes the current understanding of etiological factors and pathophysiology of sarcopenia in cirrhosis; mechanism of hyperammonemia related sarcopenia and discuss current and novel treatment options and potential avenues for future research.
CAUSES OF SARCOPENIA IN CIRRHOSIS
Sarcopenia in liver disease is multifactorial. The reasons may be related to decreased nutrition intake, malabsorption, altered metabolism, hormonal factors, hyperammonemia, and increase muscle loss.
Impaired nutrient intake and malabsorption
Poor oral intake in common in patients with cirrhosis [19]. This may be due to anorexia induced by elevated inflammatory cytokines; early satiety caused by raised intra-abdominal pressure due to ascites and altered taste due to dietary salt restrictions [20]. Impaired intestinal motility and small intestinal bacterial overgrowth in cirrhosis contribute to nutrient malabsorption [21]. Reduced bile flow in cirrhosis also contributes to fats and fat-soluble vitamins malabsorption. Pancreatic insufficiency in alcoholic related liver disease also contributes to steatorrhoea and fat malabsorption [22].
Altered metabolism
Cirrhosis is a hypermetabolic state, with increasing demand for calories and protein [23]. Hepatic glycogen stores are limited. Therefore, even after a short period of fasting, there is inappropriate excessive utilization of body fat and protein (from muscles) for gluconeogenesis [24]. This state of accelerated starvation response in cirrhosis contributes to malnutrition and sarcopenia. Increased utilization of amino acids for gluconeogenesis in cirrhosis results in low plasma branched-chain amino acids (BCAAs) levels [25]. Since BCAAs are the preferential energy source for skeletal muscles, low plasma BCAA may lead to muscle degradation [26].
Hormone deficiency
More than 90% of advanced cirrhosis has low testosterone levels [27]. Serum testosterone levels correlate with muscle mass in men [28]. Testosterone improves muscle mass and increases protein synthesis by increasing levels of insulin-like growth factor-1 (IGF-1) and consequent mammalian target of rapamycin (mTOR) activation. Testosterone also inhibits myostatin production that leads to less inhibition of muscle satellite cell activity [29]. A recent doubleblinded placebo controlled trial had demonstrated that male cirrhotics with low serum testosterone levels had marked improvement in muscle mass upon intramuscular testosterone therapy [30].
Increased muscle loss
In cirrhosis, muscle proteolysis is stimulated through the activation of the ubiquitin-proteasome activity. As a result of chronic inflammation in cirrhosis, the pro-inflammatory cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin-1, 6 are markedly elevated, which in turn stimulate muscle autophagy [31]. Moreover, autophagy induced by alcohol use directly lead to sarcopenia in patients with alcoholic liver disease [32].
Impaired muscle growth
Myostatin negatively regulates satellite cell proliferation and differentiation. Elevated myostatin levels contribute to sarcopenia in cirrhosis [33]. Hyperammonemia has been recently shown to increase muscle myostatin expression via Toll-like receptor-independent nuclear factor kappa beta activation in an animal model [34]. As serum and muscle ammonia levels are frequently elevated in cirrhosis due to impaired ureagenesis and portosystemic shunting, myostatin expression is significantly increased. The reduction in serum free testosterone levels, BCAA, and IGF-1 levels also lead to elevated myostatin levels in cirrhosis [35].
NORMAL SKELETAL MUSCLE REGULATION
The mechanistic pathways influencing muscle mass and function in cirrhosis are directly controlled by regulatory factors affecting muscle protein synthesis, differentiation and proliferation of muscle satellite cells and muscle proteolysis. Besides these, during muscle wasting, quality of muscle is also affected.
Muscle protein synthesis
Physical exercise, leucine-enriched BCAAs, testosterone, insulin and IGF-1 upregulates protein kinase B leading to mTOR activation inside the muscle cell which in turn via activation of various intracellular pathways stimulates muscle protein synthesis [36].
Satellite cell differentiation and proliferation
Myostatin, a negative regulator of satellite cells proliferation and differentiation [37] maintain the activity of satellite cells in a quiescent state within the muscle. Ammonia in muscles stimulates myostatin thereby preventing muscle growth [38].
Muscle proteolysis
Myostatin stimulates both ubiquitin-proteasome pathway (UPP) and autophagy. Hyperammonemia, inactivity and systemic inflammation also activate the UPP [32]. However, a recent study demonstrated that UPP activity is unaltered but the autophagy pathway in muscle is increased during hyperammonemia. Deranged mitochondrial function and increase in reactive oxygen species generation also activate autophagy leading to sarcopenia.
AMMONIA METABOLISM IN HEALTH AND LIVER CIRRHOSIS
Enterocytes in small intestine through the production of ammonia from glutamine and colonic microflora through the catabolism of nitrogenous sources in the form of ingested protein and secreted urea play a significant role in generating ammonia that is transported to the liver via the portal circulation [39,40].
The liver plays a major role in ammonia disposal. Periportal hepatocytes have enzymes required for urea cycle, which converts ammonia to urea to be removed by the kidney. Periportal hepatocytes also have phosphate-activated enzyme glutaminase (PAG) activity to generate glutamate needed for urea cycle. Conversely, peri-venous hepatocytes have abundant glutamine synthetase activity, which removes ammonia that has been bypassed from the periportal hepatocytes. Thus, ammonia level in blood is closely regulated by functional hepatic glutamine metabolism and urea cycle in liver [39,41].
In skeletal muscle, despite low glutamine synthetase activity, the large whole-body skeletal muscle mass makes it possible to cause significant conversion of ammonia to glutamine. The kidneys excrete amount of glutamine produced; but excess glutamine may also enter enterocytes and again produce ammonia, continuing this vicious cycle [42]. The activity of Intestinal PAG in the intestine is increased almost fourfold in cirrhotics in comparison to healthy individuals [43].
Ammonia plays a major role in the pathogenesis of minimal and overt HE in cirrhosis. Besides HE, ammonia toxicity has also been shown to affect muscles and other organs. In advanced cirrhosis and portal hypertension, loss of functional hepatocytes and bypass of portal blood flow as a result of portal-systemic shunting significantly reduce the liver capacity to remove ammonia. In addition, increased renal ammonia production also contributes to hyperammonemia [44]. Therefore, careful studies on alternative pathways of ammonia metabolism and clearance are warranted.
HYPERAMMONEMIA AND MUSCLE
In chronic liver disease, muscle plays a significant compensatory role in removing ammonia due to the presence of enzyme glutamine synthetase during the formation of glutamate to glutamine by NH3 (Fig. 1).
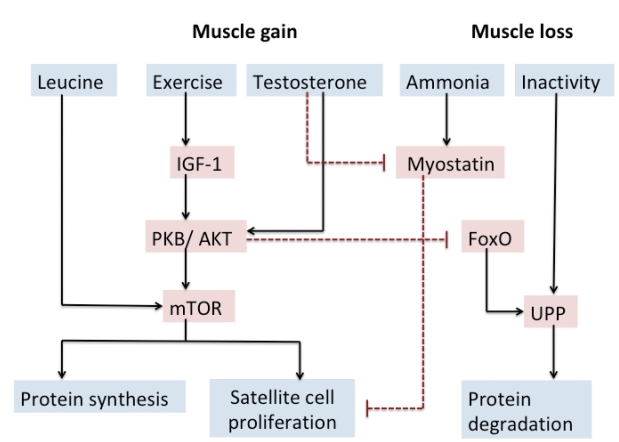
Muscle regulation and potential therapeutic strategies. Solid arrows indicate activating pathways. Dashed lines indicate inhibitory pathways. IGF-1, insulin-like growth factor 1; PKB/AKT, protein kinase B; mTOR, mammalian target of rapamycin; FoxO, forkhead box transcription factor; UPP, ubiquitin-proteasome pathway.
More recent studies by Dasarathy et al. have elegantly demonstrated that high serum and muscle ammonia level contribute to sarcopenia in cirrhosis [45]. A vicious circle develops as a cirrhotic patient develops sarcopenia as NH3 clearance decreased in sarcopenic patients, which in turn leads to a higher risk of developing hyperammonemia and HE.
Ammonia mediates myostatin transcription and expression
A previous study by Dasarathy[45] had shown increased plasma levels and increased expression of myostatin in the skeletal muscle of cirrhotic patients. Upregulation of myostatin as a result of hyperammonemia is responsible for impaired proteostasis that contributes to sarcopenia. In the skeletal muscle, ammonia enters the muscle cell, by the Rh B glycoprotein class of ammonia transporters. Hyperammonemia leads to myostatin expression via an nuclear factor of kappa light polypeptide gene enhancer in Bcells (NF-κB)-dependent toll-like receptor independent pathway. Ammonia also inhibits mTOR directly through 5’-adenosine monophosphate protein kinase dependent pathway [35]. Qiu et al. recently demonstrated that exposure of mouse skeletal muscle myotubes in culture to ammonium acetate caused a time- and concentration-dependent increase in myostatin mRNA and protein expression [34]. High concentrations of myostatin in serum were associated with sarcopenia as measured by psoas muscle index on computed tomography, high serum ammonia concentration, low serum albumin, and severity of liver disease characterized by high Child-Pugh score [46].
Ammonia mediates muscle autophagy
Autophagy has been found to be increased in muscle in experimental models of cirrhosis or during hyperammonemia but ubiquitin-mediated proteolysis is not activated [47]. Alternative possible mechanisms for activation of autophagy may include ammonia mediated mitochondrial dysfunction and generation of reactive oxygen species [48].
Ammonia-induced inhibition of muscle protein synthesis
Ammonia causes activation of general control nonderepressible 2 (GCN2) kinase (amino acid deficiency sensor) which inactivate the eukaryotic initiation factor 2 (eIF2a) and it also causes inactivation of mTORC1, resulting in global repression of mRNA translation and hence decrease in protein synthesis in the skeletal muscle [49]. Under physiological conditions, phosphorylation of eIF2a is followed by an adaptive integrated stress response (ISR) that is mediated via upregulation of activating transcription factor 4 (ATF4) which through downstream signaling pathways leads to the reversible dephosphorylation of phosphor-eIF2a [50]. The exact mechanisms and consequences of GCN2 activation during hyperammonemia and cirrhosis are not known but decreased in the plasma concentration of BCAA is a proposed mechanism of activation of GCN2.
In addition to the myostatin mediated signaling perturbations during hyperammonemia, In the mitochondria, ammonia is converted to glutamate by α ketoglutarate (α-KG), a critical tricarboxylic acid (TCA) cycle intermediate, and it then subsequently help in conversion of glutamate to glutamine in the skeletal muscle that is then exchanged for leucine by SLC7A5 [51,52]. Both hyperammonemia and loss of α-KG contribute to the sarcopenia, both loss of muscle mass and impaired contractile function due to mitochondrial dysfunction and reduced adenosine triphosphate (ATP) [53]. Recent data also show post-translational modifications of proteins is responsible for impaired muscle strength and consequent frailty [54].
However, in cirrhosis, skeletal muscle ammonia concentrations are much higher potentially favoring cataplerosis or loss of critical TCA cycle intermediate like α-KG. This results in a lower flux of the TCA cycle, impaired mitochondrial function and decreased ATP synthesis [55]. Low ATP concentrations may also cause reduced protein synthesis by hampering translation initiation.
AMMONIA AND HE
Risk of hyperammonemia increases with an increase in loss of muscle mass along with liver dysfunction as muscle helps in NH3 disposal in multiple ways. Studies have shown that venous blood ammonia levels are higher with patients with sarcopenia [56]. Since ammonia easily crosses the blood-brain barrier, neurotoxic levels of ammonia in brain astrocytes causes the development and/or persistence of HE [57]. Therefore prevalence of overt and covert HE is higher in patients with muscle depletion and decreased muscle strength [14]. Sarcopenia and frailty are thus important predictors of HE and are often under-recognized comorbid conditions (Table 2) [14,58-64].

The impact of sarcopenia and frailty on the risk of HE
NH3 in the brain acts as an osmotic agent by forming glutamine from glutamate with the help of enzyme glutamine synthetase [65]. It causes astrocyte cell swelling, which leads to cerebral edema, increases intracranial pressure, and the risk of brain herniation [66]. Astrocyte dysfunction also implicated in an increase in activity of the gamma-amino-n-butyric acid (GABA) system resulting in a decrease of energy delivery to other neural cells and the subsequent production of endogenous benzodiazepines. There are many other factors implicated in astrocyte swelling independently of hyperammonemia. These factors are benzodiazepines, hyponatremia, and inflammatory cytokines [67,68].
TREATMENT
There is no effective therapy for sarcopenia in cirrhosis. Data on treatment options aimed at mechanistic pathways that lead to sarcopenia are limited (Fig. 2). The focus of treatment is to reduce the severity and frequency of sarcopenia in cirrhosis to reduce mortality and associated complications.
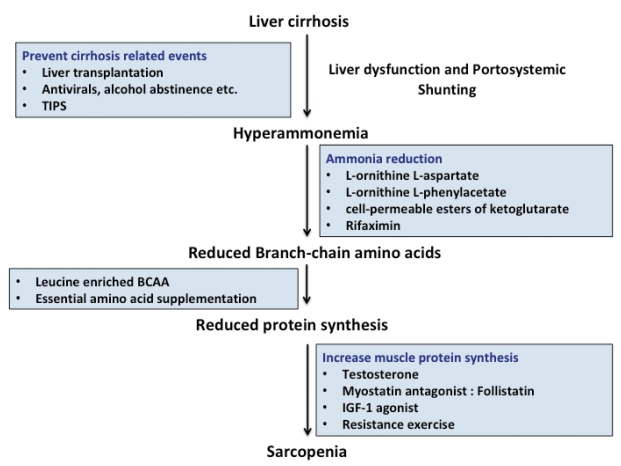
Overview of strategies to reverse sarcopenia in cirrhosis. The step-wise approach for management of sarcopenia are depicted. TIPS, transjugular intrahepatic portosystemic shunt; BCAA, branched-chain amino acid; IGF-1, insulin-like growth factor-1.
Exercise
Lack of physical exercise is linked with sarcopenia and poor clinical outcomes in cirrhotics; however, there is limited evidence that exercise can reverse sarcopenia in cirrhosis [69]. Six to 12 weeks of monitored aerobic exercise training programmes have been shown to be associated with significant improvements in muscle mass, muscle strength, peak VO2, aerobic endurance, quality of life and hepatic venous pressure gradient (HVPG) reduction [70]. Supervised physical training also prevents worsening of sarcopenia.
Both resistance and cardiovascular training exercise programs have shown benefit compared to placebo [71]. Resistance exercises improve muscle mass. It promotes skeletal muscle regeneration by inducing muscle injury [72]. Similarly, endurance exercises are important to improve functional capacity but have not been shown to do reverse sarcopenia. Future trials in sarcopenia in cirrhosis with a combination of resistance and endurance exercise may potentially improve muscle mass and strength. A study by Jones et al. have shown improvement in exercise capacity and muscle strength in response to exercise in cirrhotics [73]. However, excessive exercise increases energy expenditure and may accelerate protein catabolism. It is advisable to recommend brisk walking at least 120 to 150 minutes per week, and lifting lightweights two to three times per week.
Dietary interventions
Nutritional management plays an important role in the management of HE and possibly sarcopenia and frailty. Key nutritional strategies include; small frequent meals by shortening the time interval between meals so as to increase total caloric and protein intake. This may lead to a decrease in gluconeogenesis and increase muscle mass and strength. Simply increasing caloric intake does not delay or prevent the development of sarcopenia in cirrhosis [74]. Fat and protein catabolism is also reduced by nighttime snacks, it also prevents hyperammonemia and HE [75]. Data of nutritional supplementation for reversal of sarcopenia is limited. Cirrhotics have high protein requirements for muscle protein synthesis and maintenance or restoration of muscle mass.
Anti-ammonia measures
Treatment strategies aimed at ammonia reduction are effective in the prevention and treatment of HE but data on improvement in muscle mass and whether lowering ammonia restores proteostasis is limited.
BCAA
BCAA levels are low in cirrhosis. BCAA is highly metabolized in muscles. Supplementation of leucine-enriched BCAA provides the substrate for energy production as BCAA can replenish α-KG, which is depleted during hyperammonemia, as α-KG is aminated to glutamate. BCAA is also capable for increasing muscle protein synthesis via activation of the mTOR pathway [76].
A trial by Tsien et al. recently showed that administration of leucine-enriched BCAA in human muscle significantly reduced muscle autophagy and improved mTOR signaling that leads to increased muscle protein synthesis [26]. After three months of daily supplementation of BCAAs mixture (0.24 g/kg; 50% L-leucine, 25% isoleucine and 25% valine) in cirrhosis, they have reported reduction in blood ammonia levels and subsequent improvement in HE [27]. A recent Cochrane review also concluded that administration of BCAA improves overt HE [77], however, whether BCAA therapy improves sarcopenia need to be specially assessed in a randomized trial.
L-ornithine L-aspartate (LOLA)
A recent study by Kumar et al. demonstrated that ammonialowering therapy with LOLA given for 4 weeks in portocaval anastomosis rats significantly decreased plasma and muscle ammonia concentrations that lead to improved lean body mass, grip strength, skeletal muscle mass, and diameter compared to untreated portocaval anastomosis rats [78]. Moreover, ammonia-lowering therapy reversed elevated increased myostatin expression and autophagy markers in skeletal muscles. Further studies on longerterm and/or alternative ammonia-disposal treatments like ornithine phenylacetate and cell-permeable esters of α-KG are needed to demonstrate complete normalization of muscle ammonia concentrations [79].
Rifaximin
Non-absorbable antibiotics like rifaximin lower ammonia concentration and therefore may negatively regulate myostatin expression and may also release more BCAAs for use as an energy substrate for muscles [80].
Testosterone
The effects of testosterone therapy in cirrhosis with respect to sarcopenia has not been evaluated extensively [81,82], however a recent trial by Sinclair et al. have demonstrated that testosterone therapy in cirrhotic men with low baseline testosterone levels is safe and it increases muscle mass, bone mineral mass and reduces total fat mass [30].
IGF-1 and myostatin blockers
Follistatin, a myostatin antagonist, has been shown to improve skeletal muscle mass in animal studies. Follistatin treated portocaval anastomosis rats had significantly greater weight gain, gastrocnemius muscle size, and increased grip strength [83,84]. IGF-1 increase muscle protein synthesis and is previously reported to improve nitrogen retention in cirrhotic rats as well as reduce myostatin and increase muscle mass [85].
Reversal of portal hypertension
Transjugular intrahepatic portosystemic shunt (TIPS) placement reverses portal hypertension-related complications like ascites and variceal rebleeding. TIPS also have been suggested as a potential treatment for sarcopenia in cirrhosis. In a study of TIPS outcomes in cirrhosis, muscle mass improved post-TIPS in ≤70% of patients with a significantly reduced 12 m mortality. Mechanism of TIPSrelated reversal of sarcopenia includes the reduction in portal pressure, reduction in ascites leading to reduced metabolic rate, increased appetite, and improved nutrition [18]. In a recent study by Nardelli et al., sarcopenia constitutes a strong and independent risk factor for the development of HE after TIPS placement [86]. The mechanism for sarcopenia and HE post TIPS is likely related to reduce the capacity on the capacity of ammonia removal in presence of muscle wasting. Consequently, improvement in muscle mass and nutritional status before TIPS is warranted to decrease the incidence of HE.
Liver transplantation
Liver transplantation definitely reverses portal hypertension and hepatocyte functions but sarcopenia does not reverse and may actually worsen [19]. All patients post-transplantation require immunosuppressive medications such as corticosteroids, calcineurin inhibitors, and mTOR inhibitors and these medications are known to adversely affect muscle mass [87]. The reversibility of hyperammonemia induced signaling responses and impaired protein synthesis after liver transplantation is not known. Further longer-term follow-up studies are required to better answer this issue [88].
CONCLUSION
Sarcopenia and frailty are frequent in cirrhosis and predict important clinical outcomes, including mortality. Hepatic clearance of ammonia is significantly decreased in cirrhosis due to impaired urea synthesis and portosystemic shunts that lead to significant dependence on skeletal muscle for ammonia detoxification. The molecular pathways and mechanisms for hyperammonemia-induced sarcopenia have been recently proposed. Preclinical studies targeting hyperammonemia lay the foundation for mechanistic treatment of sarcopenia in liver disease. Strategies to improve muscle mass in cirrhosis may improve or prevent HE.
Notes
Author contributions
AJ participated in concept design, writing and critical approval of manuscript. RKJ participated in writing the manuscript and preparing tables and figures.
All authors participated in writing and final approval of manuscript.
Conflicts of Interest No financial conflict of interest.
Abbreviations
ATF4
activating transcription factor 4
ATP
adenosine triphosphate
a-KG
α ketoglutarate
BCAAs
branched-chain amino acids
eIF2a
eukaryotic initiation factor 2
GABA
gamma-amino-n-butyric acid
GCN2
general control nonderepressible 2
HE
hepatic encephalopathy
HVPG
hepatic venous pressure gradient
IGF-1
insulin-like growth factor-1
ISR
integrated stress response
LOLA
L-ornithine L-aspartate
MELD
model for end-stage liver disease
mTOR
mammalian target of rapamycin
PAG
phosphate-activated enzyme glutaminase
TIPS
transjugular intrahepatic portosystemic shunt
TNF-α
tumor necrosis factor-alpha
UPP
ubiquitin-proteasome pathway