Hyperammonemic encephalopathy: An unusual presentation of fibrolamellar hepatocellular carcinoma
Article information

Dear Editor,
Fibrolamellar hepatocellular carcinoma (FLHCC) is a rare tumor, historically thought to be a form of primary hepatocellular carcinoma (HCC). FLHCC affects primarily younger individuals (5–35 years of age), however there appears to be a double peak of incidence at ages 5–30 and 70–79 [1]. In contrast to primary HCC, the individuals affected with FLHCC classically lack a history of underlying liver disease or cirrhosis [1]. The incidence of this rare entity varies geographically, while it represents less than 1% of primary liver tumors in the United States, in Mexico it represents 5.8% of all primary liver cancers [1].
Currently, curative surgical resection remains the primary treatment modality for FLHCC. Unfortunately, there are limited therapeutic options for patients who are afflicted with unresectable or metastatic disease. From a clinical standpoint there has been limited success in the management of advanced unresectable cancers with cytotoxic chemotherapeutic regimens and the outcomes remain poor [1,2].
Similar to other abdominal tumors, FLHCC may present with nausea, abdominal fullness, abdominal pain and/or weight loss. A distinguishing feature however, is the increased rate of paraneoplastic manifestations. There have been reports of FLHCC presenting with tumoral hormone production, such as androgen aromatization resulting in gynecomastia, thyroid hormone and β-HCG production [3,4].
Hyperammonemia is a rare and dreaded complication of chemotherapy for certain malignancies, including gastrointestinal stromal tumors, neuroendocrine tumors and myeloma. Several cases have been recently reported of hyperammonemia associated with FLHCC, even in the absence of recent chemotherapy. Here we present a case of aggressive FLHCC presenting with hyperammonemic encephalopathy, in the absence of underlying liver dysfunction.
A 32-year-old female with metastatic FLHCC presented with elevated transaminases thought to be secondary to nivolumab-associated hepatitis. She was diagnosed with FLHCC at the age of 26 and initially treated with 2 stages of chemoembolization, followed by surgical removal of her remaining cancer 6 months later. Given recurrent metastatic disease, involving the gallbladder, mediastinal lymph nodes and pleura (Fig. 1), the patient received different regimens of systemic chemotherapy. Sorafenib, Ponatinib, Gemcitabine, Oxiplatin and Folfox were administered sequentially in the listed order, starting 4 years prior to presentation and discontinued about a year before symptoms started. The last chemotherapeutic intervention was nivolumab given every 2 weeks at a standard dose for 3 months and discontinued 2 months prior to presentation. The patient had mild to moderate transaminitis in the setting of Nivolumab use and this was discontinued. Additionally, interval cross-sectional imaging revealed that disease progression. The patient received dexamethasone around this time for transaminitis and persistent nausea.
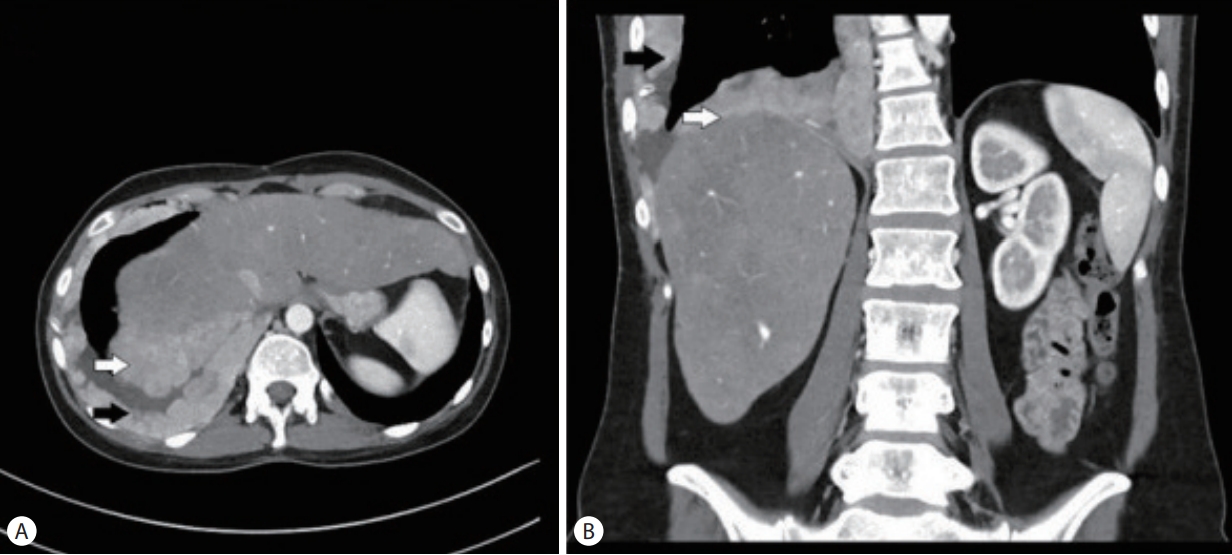
(A, B) Contrast enhanced chest computed tomography imaging showing the tumor (white arrow) and pleural metastasis (black arrow).
Approximately 10 days later, patient was hospitalized at a local hospital for acute confusion with an elevated ammonia level of 204 μmol/L. Lactulose was initiated with improvement. Her liver enzymes remained elevated although her bilirubin, alkaline phosphatase, prothrombin time and serum albumin levels remained within normal limits, indicating preserved synthetic liver function. Approximately a month later, her symptoms recurred and she was transferred to our facility. Lactulose was continued and Rifaximin was added (Fig. 2). Viral hepatitis serologies, CMV PCR and HSV were negative. An ultrasound guided liver biopsy was performed and it was negative for drug induced hepatitis or other etiologies, but it revealed mild steatosis, presumably secondary to the glucocorticoid treatment. Repeat contrast-enhanced abdominal imaging was negative for portosystemic shunts or additional findings. Plasma amino acids elevated glutamine with significant decrease in her essential amino acids which was reflective of hyperammonemia and severe catabolic stress. However, arginine and citrulline were not significantly decreased, ruling out primary inborn error of urea metabolism. Furthermore, genetic testing was obtained and it was negative for urea cycle disorders.

Time-course of ammonia levels during treatment and associated interventions. PO, per os.
Unfortunately, the patient’s mental status continued to deteriorate and it progressed to West Haven grade 4 encephalopathy, requiring intubation for airway protection. Her ammonia level at this point was 300 μmol/L, despite lactulose and rifaximin, and a decision was then made to commence ammonia scavenging therapy with sodium phenylacetate 55 mL/m2 along with 120 mL arginine and start D10W continuous infusion to avoid protein catabolism. Repeat embolization of the largest hepatic tumor was performed for possible intratumoral portosystemic shunting, but with no benefit. Subsequently to the patient was transitioned to glycerol phenyl butyrate 2.5 mL TID and 3 g citrulline q.i.d. The ammonia levels decreased, reaching a nadir of 57 μmol/L, but rose again following a single dose of prednisone for a truncal rash. Steroids were withheld as they were thought to be contributing to the elevated ammonia levels by inducing a catabolic state. The ammonia level further decreased and her mental status returned to baseline. Patient was dismissed home on L-citrulline, glycerol phenylbutyrate, lactulose and rifaximin. Unfortunately her course continued to fluctuate with multiple readmissions for recurrent encephalopathy. Given progression of her malignancy and extremely poor quality of life, the patient and the family opted to transition to comfort measures and hospice. The patient deceased several weeks later.
Although most frequently seen in advanced liver disease, portosystemic shunting and urea cycle disorders, the potential causes of hyperammonemic encephalopathy (HAE) are many. As such, a thorough evaluation and avoiding premature closure are important.
There have been reports of HAE occurring as a complication or rather in conjunction with FLHCC. Only nine such cases have been published in the literature to this date [5-12]. The hypothesized mechanisms of hyperammonemia in FLHCC are similar to other tumors, including chemotherapy-induced tissue necrosis leading to increased nitrogen levels and overloading the urea cycle [7,11]. Additionally, a recent hypothesis has been proposed by Sulaiman and colleagues suggesting HAE as a paraneoplastic manifestation of FLHCC [11]. They speculate that FLHCC might release an inhibitor of the ornithine transcarbamylase (OTC) enzyme or that there might be increased activity of ornithine decarboxylase. An experimental study in animal models has demonstrated an inverse relationship between OTC activity and rate of growth of hepatomas. An interesting phenomenon observed in this study was a significant decrease in ornithine transcarbamylase OTC activity with induced starvation of hepatomas [13]. Another study has also demonstrated significant metabolic changes on FLHCC tumor cells, including upregulation of glycolysis and suppression of Kreb’s cycle [14]. These findings might be a step toward elucidating the possible mechanisms involved in FLHCC-related paraneoplastic phenomena. Furthermore, it is likely that the pre-existing hyperammonemia has been exacerbated by the development of a catabolic state induced by chemotherapy and the use of steroids.
Currently, there are no standard guidelines for the treatment of hyperammonemia in the setting of FLHCC. Treatment focuses on management of the acute crisis and prevention of future recurrences. A three step approach is recommended: reduction of nitrogen load by avoiding catabolic states, removing excess ammonia and correcting precipitating causes. Chapuy et al. [8] have proposed an algorithm for the diagnosis and treatment of hyperammonemia in the setting of FLHCC. However, further research is required to validate these recommendations in the management of this condition.
In summary, we report a severe case of FLHCC-related hyperammonemic encephalopathy exacerbated by corticosteroid use, with only partial response to standard therapy, including ammonia scavengers. We hope with this case to increase awareness of this severe complication in patients with FLHCC.
Notes
Authors’ contribution
Douglas A.Simonetto was involved in direct patient care.
Both the authors, Douglas A.Simonetto and Nimish Thakral were involved in literature search, manuscript writing and development of hypothesis of the paraneoplastic phenomena.
Conflicts of Interest
The authors have no conflicts to disclose.
Abbreviations
FLHCC
fibrolamellar hepatocellular carcinoma
HAE
hyperammonemic encephalopathy
HCC
hepatocellular carcinoma
OTC
ornithine transcarbamylase