Role of cytoglobin, a novel radical scavenger, in stellate cell activation and hepatic fibrosis
Article information

Abstract
Cytoglobin (Cygb), a stellate cell-specific globin, has recently drawn attention due to its association with liver fibrosis. In the livers of both humans and rodents, Cygb is expressed only in stellate cells and can be utilized as a marker to distinguish stellate cells from hepatic fibroblast-derived myofibroblasts. Loss of Cygb accelerates liver fibrosis and cancer development in mouse models of chronic liver injury including diethylnitrosamine-induced hepatocellular carcinoma, bile duct ligation-induced cholestasis, thioacetamide-induced hepatic fibrosis, and choline-deficient L-amino acid-defined diet-induced non-alcoholic steatohepatitis. This review focuses on the history of research into the role of reactive oxygen species and nitrogen species in liver fibrosis and discusses the current perception of Cygb as a novel radical scavenger with an emphasis on its role in hepatic stellate cell activation and fibrosis.
INTRODUCTION
Liver fibrosis is a wound-healing response to chronic liver diseases (CLDs) and often results in cirrhosis, liver failure, portal hypertension, and hepatocellular carcinoma (HCC) [1]. CLD has various etiologies including alcohol consumption, infectious diseases such as viral hepatitis, exposure to toxins and drugs, persistent autoimmune injury, or chronic conditions of altered metabolism. Whatever its etiology, liver fibrogenesis is a dynamic and highly integrated molecular, tissue, and cellular process that distorts the hepatic architecture, and contributes to the formation of a new biochemical environment in the liver [2].
The relevance of reactive oxygen species (ROS) to liver fibrosis was first described in 1965 by Comporti et al. [3] and Ghoshal and Recknagel [4] who reported that liver injury was induced by carbon tetrachloride (CCl4) via lipid peroxidation. Two years later, Di Luzio and Hartman [5] implicated lipid peroxidation in the pathogenesis of ethanol-induced fatty liver. Figure 1 shows the timeline of research on the role of oxidative stress in liver diseases [6-17]. Almost 50 years have passed, and today oxidative stress is known to be related to chronic liver injury and hepatic fibrosis caused by ethanol [18,19], CCl4 [20], as well as non-alcoholic steatohepatitis (NASH) [21], nonobese patients with NASH [22], iron overload [23,24], and hepatitis C virus (HCV) [25]. Moreover, mitochondrial ROS26 mediate metabolic pathway signaling [27-29], and their changes affect the development and progression of CLD [30-32]. The fibrogenic progression of these diseases is associated with a significant decrease in, and/or impairment of, antioxidant defenses such as superoxide dismutase (SOD) 2 activity [33,34], or manganese (Mn) SOD [35].

Timeline of research into ROS in liver disease. ROS, reactive oxygen species; CCl4, carbon tetrachloride; NASH, non-alcoholic steatohepatitis.
The recently discovered cytoglobin (Cygb) [36], which has an antioxidant function, is present in hepatic stellate cells (HSCs) [37], the main cell type involved in liver fibrosis. In proteomic analyses, Kawada et al. [36] has found this Cygb protein in rat HSCs and named it as stellate cell activation-associated protein. Later, it was classified as the fourth member of the globin family in mammals, after hemoglobin (Hb), myoglobin (Mb), and neuroglobin (Ngb), and renamed as Cygb [38]. Figure 2 showed the 3D structure of these four members of globin family in human. Cygb is a 21-kDa protein consisting of 190 amino acids that shows ~25% identity with vertebrate Mb and Hb, and 16% identity with human Ngb [39]. Moreover, some key residues in the ligand-binding reaction are highly conserved among different species of Ngb, Mb, and Hb. By contrast, both Cygb and Ngb have unusual features, which are different from traditional pentacoordinated globins such as Mb and Hb. Spectroscopic studies have shown that Cygb and Ngb contain a hexacoordinated heme iron, to which two his imidazole groups are bound directly in both the deoxyferrous and ferric states [40]. Therefore, exogenous ligands, such as O2 or carbon monoxide, can bind to the iron after displacement of one of his imidazole groups from the axial coordination site [39,41]. However, like Mb, Cygb exhibits high intrinsic affinity for O2 [39,40]. Cygb contains two cysteines, which might form intramolecular or intermolecular disulfide bridges [42,43]. Their substitution or reduction diminish Cygb affinity to O2. This indicates that the cellular redox state influences protein structure by S-S bond formation or cleavage, thus affecting O2 binding.
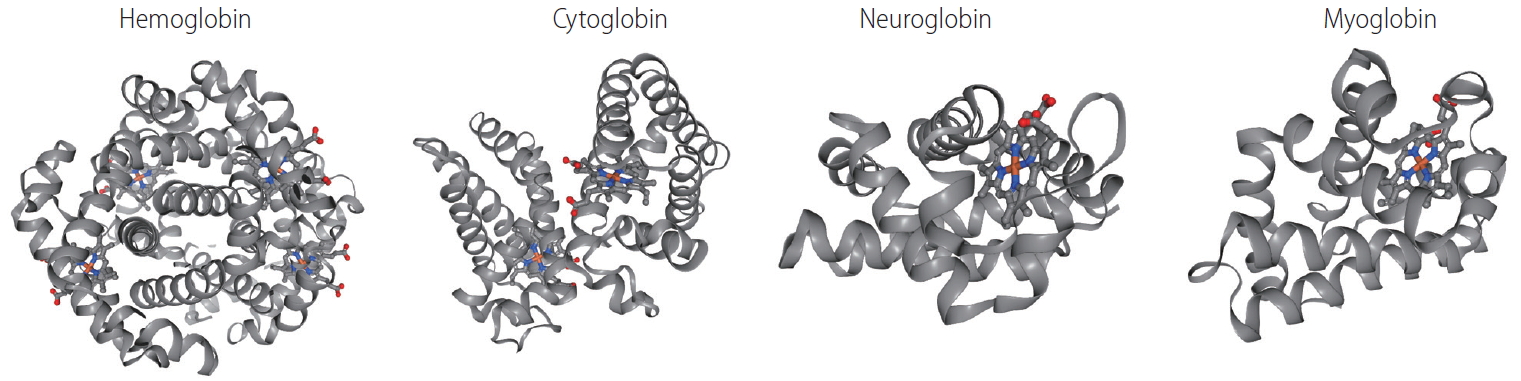
3D structure of four members of globin family. Human haemoglobin (RCSB Protein Data Bank accession number: 2HHB, tetramer [158], cytoglobin (1V5H, dimer [40], neuroglobin (1OJ6, monomer [159], and myoglobin (3RGK, monomer [160] are shown with alpha-helices and heme group. Color scheme for the elements: grey for carbon, red for oxygen, blue nitrogen, yellow for sulfur.
Not only structural but also functional and pathophysiological characterizations of Cygb have been reported. This review highlights the protective role of Cygb in liver fibrosis via its ROS-scavenging function and discusses its future prospects.
CYGB: TISSUE AND CELLULAR DISTRIBUTION
Cygb is expressed ubiquitously in all vertebrate organs, including the brain, liver, heart, lung, retina, gut, and esophagus. The expression of Cygb in mouse organs was shown in Figure 3. Looking at the liver, Cygb is detected in HSCs [36], but not in hepatocytes, Kupffer cells (KCs), endothelial cells, or myofibroblasts [37]. It is also present in the stromal cells of the red pulp of the spleen. In the kidney, Cygb expression is present in stromal cells along the proximal and distal uriniferous tubules [44]. It is expressed in fibroblasts, but not in cardiomyocytes in the heart. In the lung, it is found in chondroblasts or stromal cells along alveolar walls. In the thymus, it is present in stromal cells. Interestingly, Cygb is also found in adipose tissue (Fig. 3). A novel expression site is in melanocytes; its absence is associated with the melanocyte-to-melanoma transition [45].
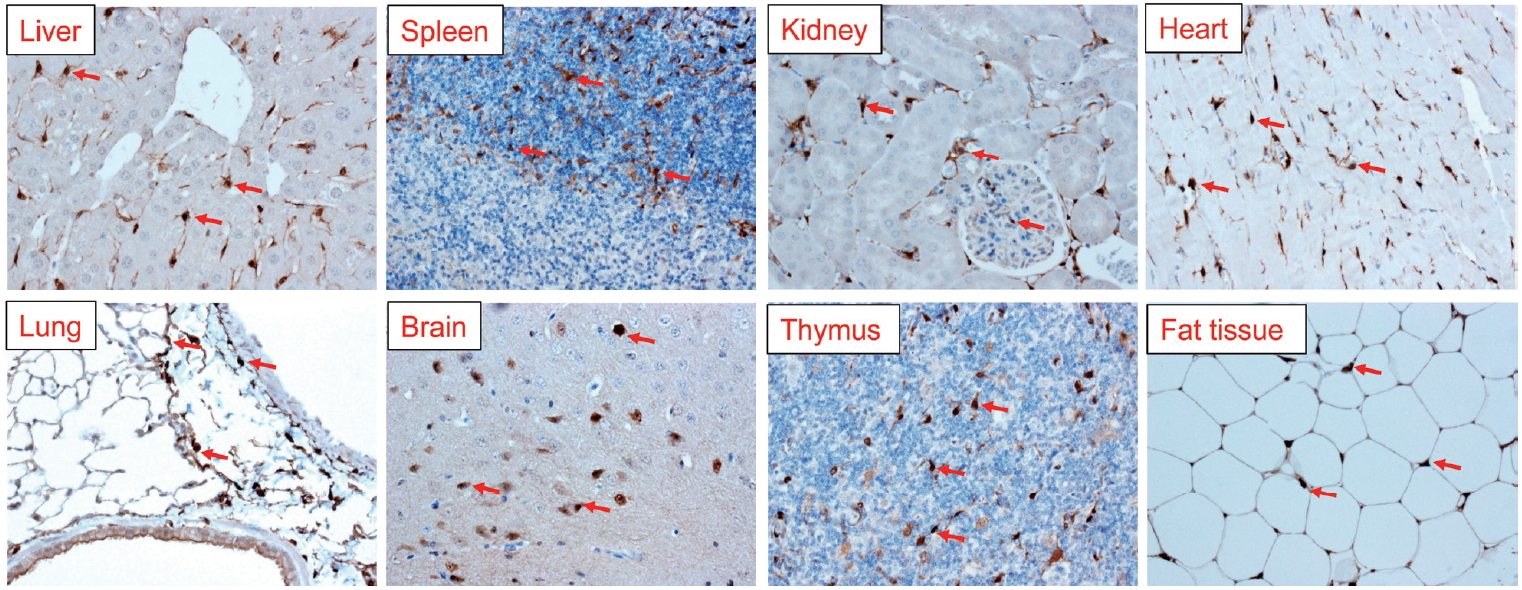
At the cellular level, Cygb is mainly expressed in fibroblast-related cell lineages such as HSCs, chondroblasts, and osteoblasts [37,46-52]. Moreover, it has been found in distinct neuron populations [47,50,53]. Cytoplasmic and nuclear Cygb localization mainly occurs in neurons. Specific Cygb expression in neurons suggests that the globin may play different roles in this cell population compared to mesenchymal cells [47]. Some studies have demonstrated that Cygb localizes in macrophages, muscles [54], hepatocytes [53], and epithelium [53-55]. However, the discrepancies between the studies on the cell-type and subcellular localization of Cygb might have arisen due to technical issues related to the specificity of the antibodies used, immunodetection methods applied, and endogenous Cygb expression level [56].
CYGB: A NOVEL RADICAL SCAVENGER
Impact of radicals in liver fibrosis
Oxygen free radicals, more generally known as ROS, along with reactive nitrogen species (RNS) are the most important radical species generated in living systems [57]. ROS and RNS play an important role in the establishment of fibrosis and subsequently in cirrhosis [58]. In a previous study, human HSCs cultured with human neutrophils stimulated to produce ROS showed evidence of not only oxidative stress but also increased procollagen α1(I) mRNA and protein levels compared to those co-cultured with unstimulated neutrophils [59]. These effects were inhibited by adding vitamin E or SOD to the culture. By contrast, the intra- and extracellular hydrogen peroxide, lipid peroxidation, and collagen type I levels were increased in HSCs co-cultured with HepG2 cells, which express cytochrome P450 2E1 (CYP2E1). In another study, an increase in collagen production was prevented by antioxidants and a CYP2E1 inhibitor [60]. Similarly, co-culture with KCs results in activation and proliferation of HSCs, along with increased production of collagen type I and hydrogen peroxide [61]. Thus, induction of oxidative stress in inflammatory cells, macrophages, and CYP2E1-positive hepatoma cells leads to the production of pro-fibrogenic mediators. In patients with alcoholic liver disease and NASH, CYP2E1 plays a critical role in ROS generation [62] and CYP2E1 is induced by alcohol [63]. One of the reasons for the increase in the CYP2E1 protein level during chronic ethanol intake is decreased proteasomal degradation [64]. A newly discovered host marker of oxidative stress, 3β-hydroxysterol Δ24-reductase, an HCV-induced oxidative stress responsive protein [65,66], may play a critical role in the pathogenesis of chronic HCV infection and associated liver diseases when aberrantly expressed [67,68]. Thus, irrespective of etiology, generation of ROS and other free radicals cause oxidative stress in chronic liver injury-induced fibrosis.
Nitrosative stress is a condition in which production of RNS is excessive, exceeding the system’s ability to neutralize and eliminate them [69]. Nitrosative stress acts together with oxidative stress to damage cells. The reaction of nitric oxide (NO) with the superoxide anion (O2–) results in formation of peroxynitrite (ONOO–) [70,71], and nitrotyrosine which in turn cause nucleotide modifications in DNA and induce dysfunction and degradation of several functional proteins [72]. In a study that used an animal model of endotoxemia, plasma S-nitrosothiol and hepatic nitrotyrosine levels were significantly higher in rats with cirrhosis than in control rats, and there was strong positive staining for nitrotyrosine in immunohistochemistry analyses of the livers of rats with cirrhosis [73]. Large amount of NO can be generated by inducible NO synthase (iNOS) in the liver, via immunological stimuli, such as bacterial lipopolysaccharide (LPS) and inflammatory cytokines, which implicated in many liver diseases, including liver fibrosis [74]. In human, Koruk et al. [75] reported that the elevated serum NO level (determination by the stable end products of NO radical, nitrite [NO2–] and nitrate [NO3–]) in patients with hepatic cirrhosis suggesting NO contributes to the progression of cirrhosis. The levels of NO derivatives, as mentioned above, nitrotyrosine, and nitrosothiols, are also increased in autoimmune hepatitis [76], and primary biliary cholangitis [77,78]. In NASH patients, both iNOS and nitrotyrosine levels are significantly elevated [79]. These findings suggest a major role for NO in chronic liver injury-associated fibrogenesis.
A causative role for oxidative stress in liver fibrogenesis has been strongly suggested by several reports that supplementation with antioxidants prevents fibrogenic progression. Experimental models of liver fibrosis/cirrhosis have been used to evaluate antioxidant compounds such as polyunsaturated phosphatidylcholine [80], peroxisome proliferator-activated receptor (PPAR) α ligand [81], ursodeoxycholic acid [82], and resveratrol [83-86]. Clinically, the only drug used to treat acetaminophen overdose patients is the precursor of glutathione (GSH), N-acetylcysteine [87]. Other antioxidant treatments include GSH, resveratrol, or Mito-TEMPO, a Mn superoxide dismutase mimetic [88,89]. S-adenosylmethionine, silymarin, and vitamin E have been tested in liver fibrosis/cirrhosis patients. An effect of vitamin E has been reported in ASH- or NASH-induced fibrosis, where histological findings such as steatosis, inflammation, and fibrosis were improved [90]. The most promising results were reported from PIVENS trial [91], which was performed in 247 patients for 96 months. Vitamin E treatment led to clear histological regression without fibrosis progression. Currently, the ClinicalTrials.gov website lists 14 early and phase I–IV clinical trials of antioxidant therapies for liver cirrhosis. In these studies, vitamins, particularly vitamin E, are the most frequently studied antioxidants used as dietary supplements.
CYGB scavenges ROS and RNS
Cygb holds intrinsic O2-binding capacity
As a member of the globin family, the heme iron of Cygb has the same affinities for exogenous ligands and the same equilibrium constant for oxygen as Mb [36,39]. Cygb contributes to intracellular O2 supply [92], acting as an O2 reservoir or as a signal transducer in O2-sensing pathways [53,93]. A change in the cellular redox state could promote conformational changes in Cygb and increase O2 release (e.g., the reduction of S-S bridges by reducing agents H+ or nicotinamide adenine dinucleotide) [42].
Cygb scavenges ROS
Cygb has several roles, including detoxification of ROS, involvement in NO metabolism, protection from apoptosis, and lipid metabolism [93-96]. Plasma-produced ROS/RNS can oxidize Cygb proteins, leading to a conformational change, thereby enabling access to the heme and facilitating ligand binding [97]. Interestingly, human Cygb mutants with one or both terminal domains truncated show slightly higher superoxide scavenging activity than wildtype Cygb [98]. Overexpression of Cygb under conditions of oxidative stress have been found in a number of studies [50,99]. Its overexpression protected human neuroblastoma SH-SY5Y cells from H2O2-induced cell death [100,101] and rescues the human neuronal cell line TE671 from pro-oxidant Ro19-8022-induced DNA damage [102]. Furthermore, in vitro and in vivo overexpression of Cygb in rat HSCs protect these cells against oxidative stress and inhibit their differentiation into an active phenotype [103]. Therefore, the question is whether oxidative stress is magnified in the absence of Cygb. We generated Cygb-knockout mice and challenged them with various factors that induce liver diseases. First, the mice were treated with 0.05 ppm diethylnitrosamine, an established liver carcinogen, for 36 weeks. Liver tumors occurred in 57.1% of the knockout mice compared to 0% of the wild-type mice. In this model, background liver tissues of knockout mice showed marked development of liver fibrosis, augmented inflammatory reactions, and overproduction of ONOO- [104]. Second, mice were given a choline-deficient L-amino acid-defined diet for 32 weeks to induce steatohepatitis. Unexpectedly, 100% of Cygb-knockout mice developed multiple liver tumors, compared to 0% of the wild-type mice. Again, background liver tissues showed development of liver fibrosis and augmented inflammatory reactions, accompanied by DNA double-strand breaks (γH2AX expression) in hepatocytes. These results suggest a protective role for Cygb against oxidative stress, liver fibrosis, and cancer development in the presence of chronic inflammation [52]. Recently, Latina et al. [105] reported that the Cygb gene is transcriptionally regulated by ΔNp63 in primary epithelial cells (keratinocytes) and in cancer cells (H226, MCF-7) under both normal proliferation conditions (normoxia) and following oxidative stress. Taken together, these reports suggest that, in addition to its function as a gas carrier, Cygb acts as a cytoprotective molecule under hypoxia and oxidative stress.
Nitric oxide scavenger
Many globins, including Cygb, show NO dioxygenase activity [106-108]. In the oxy-ferrous state, all human Ngb and Cygb, rice nsHb (riceHb1), Synechocystis Hb (cyanoglobin, SynHb), and horse heart Mb rapidly destroy NO in vitro, and Cygb has the highest consumption rate [109]. At a low O2 level (0–50 mM), Cygb with cellular reductants regulates the NO consumption rate in response to changes in O2 concentration and is around 500-fold more sensitive to changes in the O2 level than Mb [110]. Indeed, the NO dioxygenase activity of Cygb is rapid with or without a disulfide bond; however, binding of the distal histidine following dissociation of the nitrate is affected by the presence or absence of the disulfide bond [111]. The NO scavenging function of Cygb protects the NO-sensitive aconitase, and decreases ONOO– formation [108]. Cygb plays a critical role in the regulation of vascular tone and blood pressure via NO metabolism [94]. Importantly, CYGB is expressed in vessels primarily in differentiated medial vascular smooth muscle cells, where it regulates neointima formation and inhibits apoptosis after injury [112]. Moreover, when the Cygb-KO mice were challenged with bile duct ligation (BDL) induced liver cholestasis, liver injuries including hepatocyte damage, oxidative stress, and fibrosis were massively developed compared to corresponding WT (Fig. 4). This severe liver cholestasis is accompanied by markedly increased apoptosis cell dead as indicated in Figure 5. Furthermore, the levels of nitrite and nitrate in the serum, urine, and liver in Cygb-deficient mice are all significantly elevated [113]. Interestingly, treatment of NO inhibitor to BDL-treated Cygb-KO mice can ameliorate this cholestasis condition [113]. Thus, the NO-scavenging function of Cygb is crucial for protecting cells/tissues from NO accumulation.
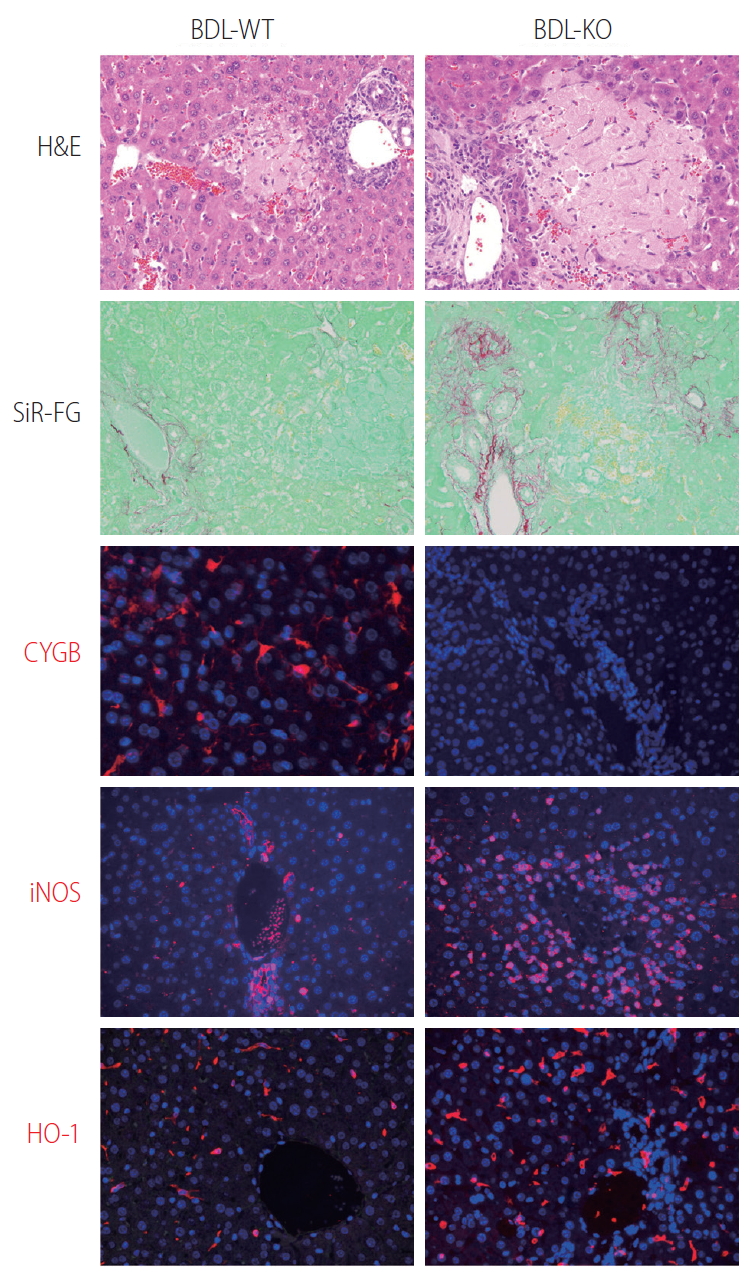
Loss of Cygb promoted bile duct ligation induced liver cholestasis. Bile duct ligation was performed in WT (BDL-WT) and CygbKO (BDL-KO) mice. Liver tissues from 1 week of BDL stained with H&E, and Sirius-Red and Fast Green (SiR-FG) showed marked hepatocyte damage and severe fibrosis in KO compared with WT mice. Immunofluorescent staining of Cygb (red) showing the absence of Cygb in KO liver. Both iNOS and HO-1 (red), the markers of RNS and ROS, respectively, revealed strong oxidative stress took place in the KO mice after 24 hours of BDL. DAPI, blue, was used as nuclear counterstain. Original magnification, ×400. H&E, Hematoxylin and Eosin; Cygb, cytoglobin; iNOS, inducible nitric oxide synthase; RNS, reactive nitrogen species; ROS, reactive oxygen species; DAPI, 4’,6-diamidino-2-phenylindole.
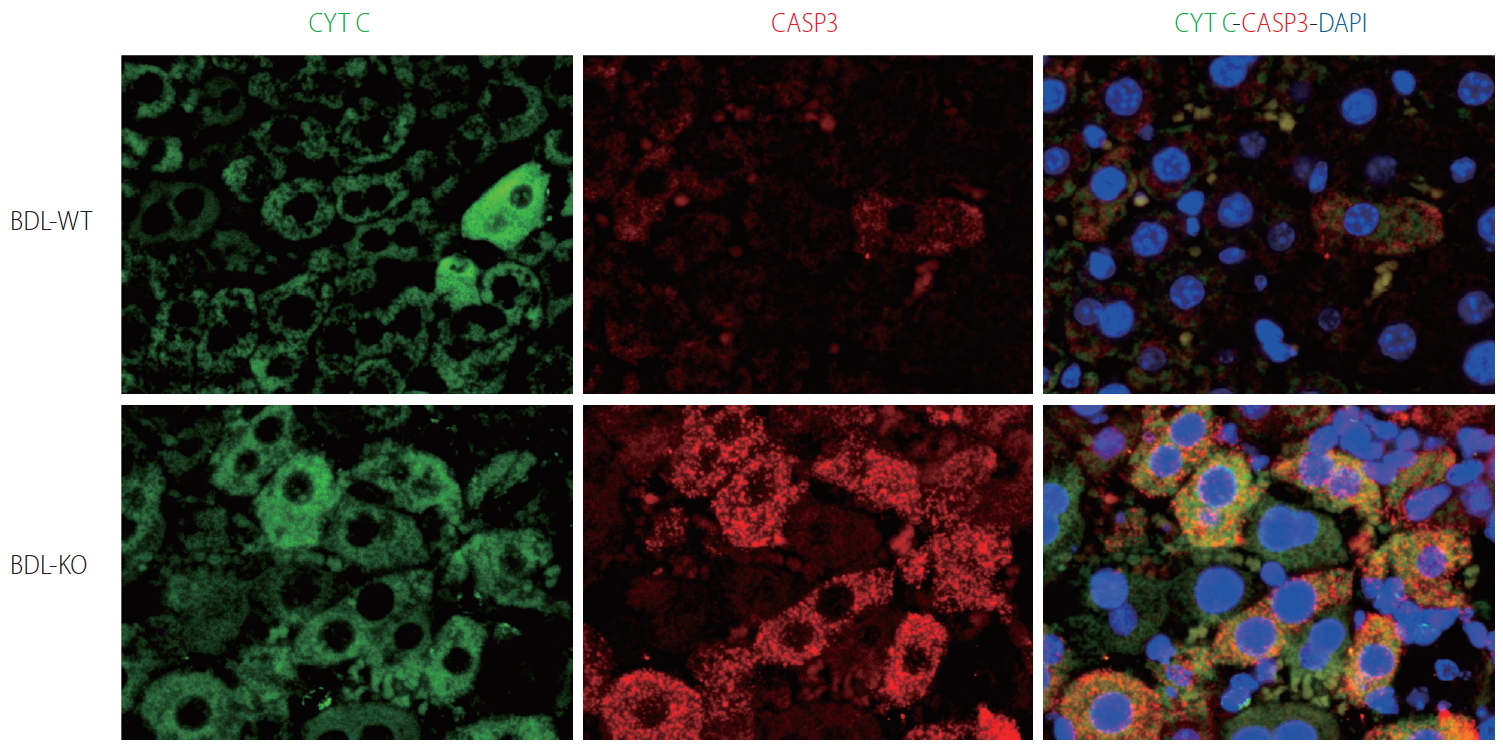
Hepatocyte apoptosis in Cygb-KO liver under BDL. Expression of markers of apoptosis cytochrome C (CYTC) (green) and active + pro Caspase (CASP) 3 (red) after 24 hours of BDL in WT and KO mice. DAPI, blue, was used as nuclear counterstain. Original magnification, ×1,200. BDL, bile duct ligation; Cygb, cytoglobin; DAPI, 4’,6-diamidino-2-phenylindole.
CYGB SUPPRESSES HSC ACTIVATION AND FIBROSIS DEVELOPMENT
HSC and hepatic fibrosis
HSCs reside in the space of Disse, between the basolateral surface of hepatocytes and the anti-luminal side of sinusoidal endothelial cells [114], and contain retinoid and lipid droplets [115]. Under physiological conditions, HSCs exhibit a quiescent phenotype and express neural markers, such as glial fibrillary acidic protein, synemin, synaptophysin [116], nerve growth factor receptor p75 [117], desmin, CD146, and hepatocyte growth factor [118]. The notion that HSCs are a major collagen-producing cell type in the normal liver was experimentally verified in 1984 by Senoo et al. [119] and in 1985 by Friedman et al. [120] Friedman also pointed out that normal HSCs exhibit not only fibroblastic characteristics but also smooth muscle cell-like features, such as the production of basement membrane collagen (type IV collagen) [120] and the expression of the intermediate filament protein desmin [121]. Beyond their role as the major collagen-producing cells, their activation is a key issue in liver fibrosis [122]. Stimuli such as oxidative stress signals (reactive oxygen intermediates), apoptotic bodies, LPS, and paracrine signals from neighboring cells including KCs, liver sinusoidal endothelial cells, and hepatocytes can trigger HSC activation or transdifferentiation into myofibroblast-like cells that acquire contractile, proinflammatory, and fibrogenic properties [1,123,124]. The dead or dying hepatocytes as well as leukocytes phagocytosing the cells release inflammatory mediators, damage-associated molecular patterns or danger signals, which initiate and perpetuate a non-infectious “sterile” inflammatory response. Among such mediators, tumor necrosis factor (TNF), interleukin (IL) 6, IL-1β, ROS, hedgehog ligands, and nucleotides contribute to the initiation of HSC activation [125-127]. Other key elements critical for the fibrotic activity in HSCs are NADPH oxidase (NOX) enzymes, in which all NOX1, NOX2, and NOX4 are upregulated in activated HSCs compared to quiescent HSCs [128,129]. Indeed, the mRNA for the cytoplasmic factor p47phox and the cell membrane proteins NOX2 and NOX1 are detected at very low levels in quiescent HSCs by real time Reverse Transcription Polymerase Chain Reaction, while they are highly expressed following HSC activation in culture and in cells freshly isolated from patients with liver fibrosis [130].
The pathways most involved in HSC activation and deactivation are related to membrane receptor signaling, including transforming growth factor beta (TGFβ), which has an autocrine positive feedback loop that drives fibrogenesis via mothers against DPP homolog (SMAD) 2/3 [131,132]; platelet-derived growth factor, a potent chemoattractant induced during initiation of HSC activation that enhances inflammatory and fibrogenic responses [133,134]; and connective tissue growth factor and epidermal growth factor receptors, which are overexpressed and phosphorylated in activated HSCs [135-138]. A recent study [139] identified transcription factors that prevent activation of HSCs and promote fibrosis resolution, including E26 transcription-specific transcription factor (EST1), ETS2, GATA binding protein (GATA4), GATA6, interferon response factor (IRF) 1, and IRF2. In particular, GATA6 and PPARγ are required for inactivation of human HSCs and regression of liver fibrosis in mice [139]. Further analyses of the metabolic changes in HSCs during the initial and chronic phases of fibrosis have provided insight into the metabolic regulation of HSC activation including metabolism of retinol [140,141], lipid [142,143], nitrogen [144], redox biology [128,145], and endoplasmic reticulum stress [146], which are important for development of targeted interventions to reverse HSC activation or trigger their apoptosis [147]. Clinical and experimental studies have demonstrated that the regression of liver fibrosis may be caused by the disappearance of activated HSCs/myofibroblasts by apoptosis [126,148], inactivation into a quiescent-like state [149,150], or senescence [151,152].
Cygb regulates HSC activation and fibrogenesis
The ROS-scavenging function of Cygb is evidenced by its ability to detoxify radicals via reaction with its heme [153]. In Xu et al. [103], overexpression of Cygb protected primary rat HSCs against oxidative stress, as assessed by reduced production of malondialdehyde and 4-hydroxynonenal, biomarkers of lipid peroxidation. In Stone et al. [154], Cygb expression was correlated with a more quiescent phenotype of stellate cells in culture and Cygb was regulated by the extracellular matrix through integrin signaling in a manner dependent on activation of focal adhesion kinase. Consistently, in human liver tissues damaged by HCV infection at various fibrosis stages, the number of Cygb-positive cells decreases with fibrosis progression [37]. Interestingly, Cygb is abundant in HSCs but absent in myofibroblasts rich in fibrotic septum and positive for α-smooth muscle actin (αSMA), fubulin-2 and Thy-1 [37]. In other fibrosis conditions, overexpression of Cygb in human tendon fibroblasts decreases the expression levels of fibronectin, collagen I, collagen III, TGF-β1 and hypoxia-inducible factor 1, suggesting an antiscarring effect of Cygb post-glaucoma surgery [155]. Randi et al. [156] demonstrated abundant Cygb expression in human podocyte lines and that it has an antioxidant effect in chronic renal diseases.
We have generated, for the first time, a new transgenic (TG) mouse line in which both Cygb and mCherry reporter gene expression is controlled under the native Cygb gene promoter. Administration of a single dose (50 mg/kg) of thioacetamide (TAA) in Cygb-TG mice resulted in lower levels of serum alanine aminotransferase and oxidative stress than those of wild-type mice. At 10 weeks of TAA administration, Cygb-TG livers exhibited reduced neutrophil accumulation, cytokine expression and fibrosis, and high levels of quiescent HSCs [157]. HSCs in the absence of Cygb (HSCsCygb-null) become enlarged with a developed αSMA network after 7 days in culture and lose cellular lipid droplets more rapidly than HSCsCygb-wild. Moreover, HSCsCygb-null shows a pre-activated phenotype with increased oxidative stress and markedly elevated expression of cytokines and chemokinse such as IL-6, TNFα, IL-1β, chemokine (C-X-C motif) ligand-1, -2, and chemokine ligand-2, -3, -4 [52]. By contrast, primary HSCs isolated from Cygb-TG mice (HSCCygb-TG) exhibit a significantly decreased αSMA, collagen 1α1, and TGF-β3 after 4 days in culture compared to WT cells. HSCsCygb-TG are resistant to H2O2-induced αSMA expression [157]. Thus, cell-specific overexpression of Cygb attenuates HSC activation and protects mice against TAA-induced liver fibrosis presumably by maintaining quiescence of HSCs. Figure 6 shows the role of Cygb in liver fibrosis.
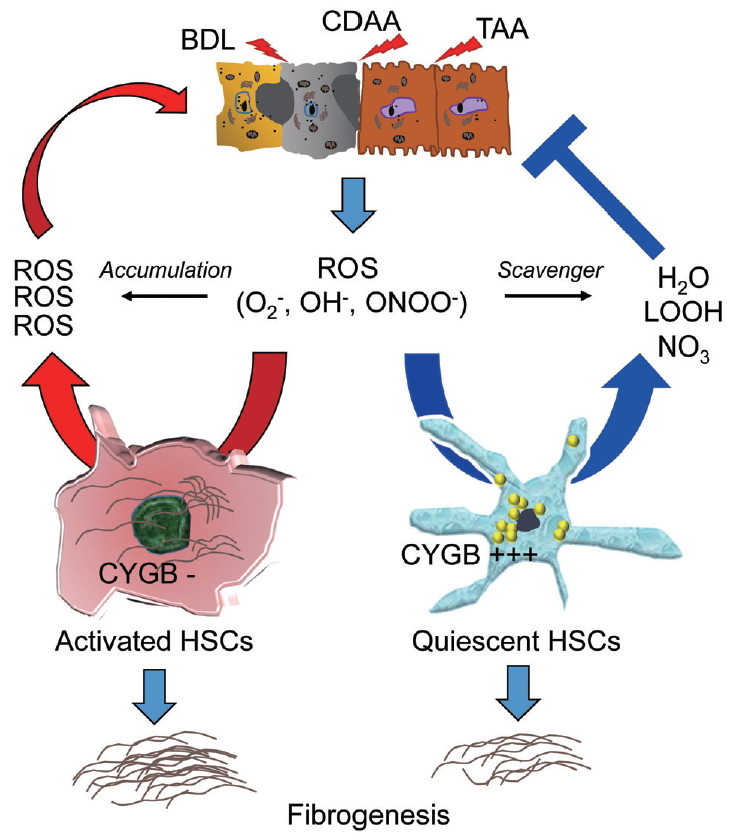
ROS-scavenging function of Cygb in liver injury. Liver injury induced by, for instance, bile duct ligation (BDL), choline deficient amino acid-define diet (CDAA), or thioacetamide (TAA) released ROS including O2–, OH–, ONOO– radicals which can induce HSCs activation, and fibrosis development. In the absence of Cygb (Cygb-), these radicals have not been scavenged, thus promote HSCs activation and magnified both liver damage and fibrosis. This cycle is suppressed when Cygb is overexpressed (Cygb+++) and scavenges ROS to H2O, LOOH, or NO3. ROS, reactive oxygen species; ONOO−, peroxynitrite; LOOH, lipid hydroperoxides; Cygb, cytoglobin; HSCs, hepatic stellate cells.
CONCLUSION AND FUTURE DIRECTION
More than 600 clinical trials of antifibrotic drugs are underway (http://www.ClinicalTrials.gov). One reason for this is the recognition of the central role of HSCs in liver fibrosis. Promising approaches to removing fibrogenic cells are being evaluated, including drug-delivery systems targeting activated HSCs. In parallel, antioxidant therapy has been targeted by the recovery of antioxidant enzymes/compounds and by reducing the production of ROS and RNS. Furthermore, a number of recent studies have explored the protective role of Cygb in hepatic fibrosis, which is mediated by inactivation of HSCs. Hopefully, the best drug for anti-fibrotic therapy will be discovered in the near future.
Notes
Authors’ contribution
LTTT, HH, and NK contributed to the literature review, and manuscript preparation.
Conflicts of Interest: The authors have no conflicts to disclose.
Acknowledgements
NK was supported by a Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (JSPS) through Grant No. J192640002 (2019-2022) and by the Japan Agency for Medical Research and Development (AMED, 2019-2022).
LTTT was supported by a Grant-in-Aid for Scientific Research from JSPS through Grant No. J192640023 (2019-2022), and by the GILEAD Science Research Scholar Program in Liver Diseases Asia (2019-2021).
Abbreviations
BDL
bile duct ligation
CCl4
carbon tetrachloride
CLD
chronic liver disease
Cygb
cytoglobin
CYP2E1
cytochrome P450 2E1
GSH
glutathione
Hb
hemoglobin
HCC
hepatocellular carcinoma
HCV
hepatitis C virus
HSCs
hepaticstellate cells
IL
interleukin
iNOS
inducible nitric oxide synthase
IRF
interferonresponse factor
KC
Kupffer cells
LPS
lipopolysaccharide
Mb
myoglobin
Mn
manganese
NASH
non-alcoholic steatohepatitis
Ngb
neuroglobin
NO
nitricoxide
NOX
NADPH oxidase
O2−
superoxide anion
ONOO−
peroxynitrite
PPAR
peroxisome proliferator-activated receptor
RNS
reactive nitrogen species
ROS
reactive oxygen species
SOD
superoxide dismutase
TAA
thioacetamide
TG
transgenic
TNF
tumor necrosis factor
αSMA
α-smooth muscle actin