Evidence-based clinical advice for nutrition and dietary weight loss strategies for the management of NAFLD and NASH
Article information

Abstract
Nonalcoholic fatty liver disease (NAFLD) is the most common cause of chronic liver disease worldwide and affects approximately one third of adults in the United States. The disease is becoming a global epidemic as a result of the rising rates of obesity and metabolic disease. Emerging data suggest weight loss of ≥10% overall body weight is beneficial in resolving steatosis and reversing fibrosis. Prospective trials comparing various diets are limited by lack of sufficient power as well as pre- and post-treatment histopathology, and therefore no specific diet is recommended at this time. In this narrative review we examine the pathophysiology behind specific macronutrient components that can either promote or reverse NAFLD to help inform more specific dietary recommendations. Overall, the data supports reducing saturated fat, refined carbohydrates, and red and processed meats in the diet, and increasing the consumption of plant-based foods. Diets that incorporate these recommendations include plant-based diets such as the Dietary Approaches to Stop Hypertension, Mediterranean, vegetarian, and vegan diets.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) encompasses a spectrum of disorders ranging from simple steatosis to steatohepatitis. NAFLD is the most common cause of chronic liver disease worldwide, affecting approximately 25% of the adult population in the United States (US) and globally [1]. Approximately 25% of patients with NAFLD have nonalcoholic steatohepatitis (NASH), which is associated with a 20% risk of progression to cirrhosis [2]. It is estimated that NAFLD affects 40–70% of patients with type 2 diabetes mellitus (T2DM) [2-4], 67% of adults with a body mass index (BMI) between 25 and 30 kg/m2, and up to 91% of adults with a BMI >30 kg/m [5-9].
The diagnosis of NAFLD requires >5% hepatic steatosis with lack of secondary cause for hepatic fat accumulation. Steatosis results from a surplus of free fatty acids (FFAs) either from excessive lipolysis (60%), de novo lipogenesis (DNL, 25%), dietary FFA (15%), diminished export by very low-density lipoprotein (VLDL), or impaired beta-oxidation (Fig. 1) [10]. Excess FFAs are stored as triglycerides in the hepatocytes. In NAFLD, peripheral lipolysis is resistant to suppression by insulin, further increasing serum FFA levels [11]. Lipotoxicity, along with oxidative stress and a pro-inflammatory environment, lead to NASH [12].

The pathogenesis of NAFLD. TG, triglycerides; FFAs, free fatty acids; SREBP-1c, sterol regulatory element-binding protein-1c; ChREBP, carbohydrate response element-binding protein; NASH, nonalcoholic steatohepatitis; NAFLD, nonalcoholic fatty liver disease; VLDL, very low density lipoprotein.
Due to lack of approved pharmacologic therapy, current treatment recommendations for NASH are for weight loss of ≥10% total body weight, which is associated with resolution of steatohepatitis and fibrosis regression [13-15]. Prospective trials comparing various diets are limited by lack of sufficient power as well as preand post-treatment histopathology [13]. As a result, the American Association for the Study of Liver Disease has not made any specific dietary recommendation for NAFLD at this time. While guidelines from the European Association for the Study of the Liver (EASL) recommend exclusion of NAFLD-promoting components (processed food, high fructose foods and beverages) in addition to a macronutrient composition in line with a Mediterranean diet, this recommendation is only supported by evidence graded as “moderate” in quality [14]. Here we review the pathophysiology behind how the quality of proteins, carbohydrates (CHOs), and fats can promote or reverse NAFLD, the available data on specific diets, and discuss ongoing knowledge gaps for future research.
ENERGY AND CALORIC RESTRICTION
Observational studies
The diabetes, obesity and NAFLD epidemics are products of a significant rise in net population energy intake, resulting from energy-dense foods and a sedentary lifestyle. A high-calorie diet is fundamentally linked to obesity and is the initial trigger point for NAFLD through adipose tissue expansion, increased inflammation and mitochondrial dysfunction. It is often imbalanced with high quantities of saturated fatty acids (SFAs), refined CHOs, sugar-sweetened beverages (SSBs) and alcohol excess. Understanding the individual impact of these factors is therefore a challenge for clinical trial design, and limits our understanding of the role of specific macronutrients outside the context of excess energy intake.
In terms of observational studies, a cohort study of 55 patients with NAFLD and 88 healthy controls observed that NAFLD patients consumed more calories overall (2,739 vs. 2,173 kcal), however the macronutrient composition (fat, CHO, fructose) was largely comparable between groups [16]. Given this, energy restriction, and thereby weight loss, plays a crucial role in the treatment of these patients. The largest prospective trial to date in this field followed 293 patients with biopsy-proven NASH for 52 weeks. Patients were advised to follow a low fat, hypocaloric diet which contained 750 kcal/day less than their daily energy needs, in addition to walking 200 minutes per week [15]. Overall 19% had fibrosis regression, 47% had reduction in NAFLD activity score (NAS) on histology and 25% achieved complete resolution of steatohepatitis. The highest rates of fibrosis regression, NAS reduction and NASH resolution occurred in patients achieving ≥10% weight loss, however benefits were also seen for weight loss of ≥5%. Using nutritional counselling, a small study of 15 patients with biopsy-proven NASH and a BMI >25 found histologic improvement to be associated with greater weight reduction [17]. After 1 year of calorie restriction (mean reduction of 195 kcal/day), nine patients had histologic improvement and six had stable NAS. Histological improvement was associated with greater changes in weight reduction.
Pathophysiology
A high-caloric diet leads to adipose tissue expansion, one of the sentinel events in the pathogenesis of NAFLD. Visceral adipose tissue (VAT) is particularly biological active. Excess accumulation of VAT increases the production of FFAs through reduced insulin sensitivity, leading to increased fatty acid influx into the liver, DNL and insulin resistance. Hypertrophy and hyperplasia of adipocytes results in hypoxemia and adipocyte dysfunction [18]. This exacerbates insulin resistance and dyslipidemia, and creates a pro-inflammatory environment within the adipose tissue. The secretion of adipokines (adiponectin, resistin, leptin, visfatin) and pro-inflammatory cytokines (interleukins and tissue necrosis factor α) from VAT induce a state of systemic low-grade chronic inflammation which can promote the onset of NAFLD and NASH [18-20].
Randomized controlled trial data
The findings from key randomized controlled trials (RCT) in this field involving lifestyle intervention for NAFLD and NASH are summarized in Table 1 [21-24]. Weight loss is consistently identified as being central to the metabolic benefits that result from calorie restriction. A meta-analysis of therapeutic options for NAFLD, including lifestyle interventions, reported that weight loss of ≥7% (achieved by <50% of patients even with intensive multi-disciplinary support) led to improved histological disease activity determined by NAS, although there was no impact on fibrosis [25].

Summary of randomized controlled trial data examining the influence of hypocaloric diets on hepatic steatosis
While guidelines currently recommend continuous energy restriction along with physical activity [26], there is increasing popular interest in intermittent fasting (IF), i.e., a low calorie period lasting less that 24 hours followed by a normal period of feeding. Evidence suggests that intermittent energy restriction results in equivalent weight loss compared to continuous energy restriction in the short-term, with a lack of long-term data [27]. Several RCTs have looked at the effectiveness of IF in the setting of NAFLD. An 8 weeks modified alternate-day calorie restriction was found to lead to reductions in BMI, liver enzymes, liver steatosis and liver stiffness (based on shear wave elastography) compared to no intervention with adherence rates of 75–83% [28]. A larger trial comparing alternate-day and time-restricted feeding with a control group for 12 weeks revealed that both diets led to significant short-term reductions in weight and improvements in dyslipidaemia, although no changes were seen for fasting levels of insulin or liver stiffness [29].
Clinical advice
Calorie restriction with a 500–1,000 kcal daily deficit is an extremely effective lifestyle intervention for both the prevention of NAFLD and histological improvement in patients with established disease. The goal of calorie reduction should be to achieve ≥10% overall body weight loss.
PROTEIN
Observational studies
The consumption of animal protein, specifically red and processed meat, is associated with higher all-cause, cardiovascular and cancer-related mortality compared to plant protein [30-41]. In a large US cohort, red and processed meats were associated with nine causes of death, with the strongest correlation being for mortality from chronic liver disease [34]. High animal protein intake is also associated with NAFLD in overweight Caucasians independent of sociodemographic, lifestyle and metabolic traits [42]. Animal protein is positively associated with high fatty liver index (FLI) scores, whereas plant protein is inversely related [43]. In a cross-sectional study, patients with NAFLD ate 27% more animal protein compared to controls (P<0.001), with 46% of those in the highest quartile of consumption having NAFLD, compared with only 17% in the lowest quartile (P=0.001) [44]. A follow-up study showed a significant association between total (P=0.028), red and/or processed meat (P=0.031) consumption with NAFLD and insulin resistance even after adjustment for BMI, physical activity, alcohol, energy, SFA and cholesterol intake [45].
Pathophysiology
Red and processed meats likely lead to NAFLD, insulin resistance and T2DM as a result of their high content of SFAs, cholesterol, heme-iron, nitrates and nitrites, preservatives, advancedglycation end-products and branched chain amino acids (BCAAs) [46]. BCAAs, found in higher concentrations in animal protein, lead to impaired insulin sensitivity by recruiting mammalian target of rapamycin (mTOR) and assembling mTOR complex 1 in combination with insulin (Fig. 2) [47]. mTOR complex 1 induces sterol regulatory element-binding protein-1c (SREBP-1c) leading to DNL [48,49]. Diets low in methionine (found predominantly in meat, fish and dairy products), can prevent the development of insulin resistance in animal models via activation of fibroblast growth factor 21 (FGF21), which inhibits SREBP-1, suppressing DNL while activating hepatic FFA oxidation [50-55]; although it should be noted that in addition to methionine-rich diets, methionine-deficient and methionine and choline-deficient diets, can also induce NAFLD in the animal model as a result of their ability to promote lipid dysregulation and oxidative stress [56,57]. Red and processed meats also contain high levels of phosphatidylcholine and L-carnitine, which are metabolized to trimethylamine (TMA) by gut microbiota (Fig. 2). TMA is oxidized in the liver by hepatic flavin-containing monooxygenases to form trimethylamine oxide (TMAO), which is then released into the circulation. TMAO promotes atherosclerosis via the up-regulation of multiple macrophage scavenger receptors [58,59], and high TMAO levels correlate with increased incidence of major cardiovascular events [60]. It is hypothesized TMAO may promote NAFLD by altering the synthesis and transport of bile acids, decreasing the overall bile acid pool and reversing the direction of cholesterol transport and glucose and energy homeostasis [61]. Indeed, plasma TMAO levels correlate with the presence and severity of biopsy proven NAFLD in a large Chinese adult population [61]. Individuals eating a vegan diet have an altered intestinal microbiota composition compared to omnivores, with reduced capacity to produce TMAO [62].
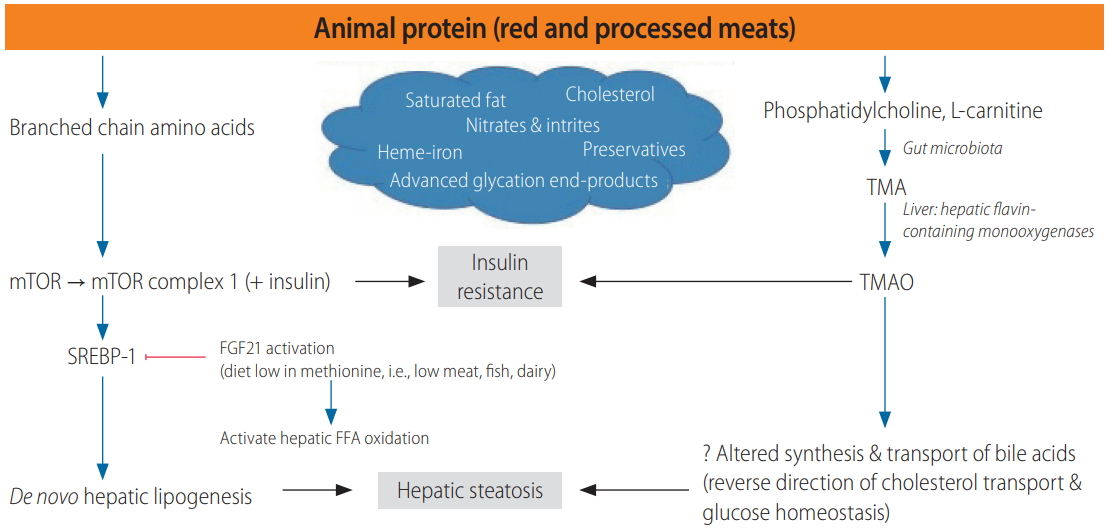
Potential mechanisms linking the consumption of animal protein with the development of NAFLD. TMA, trimethylamine; mTOR, mammalian target of rapamycin; TMAO, trimethylamine oxide; SREBP-1, sterol regulatory element-binding transcription factor 1c; FGF21, fibroblast growth factor 21; FFA, free fatty acid; NAFLD, nonalcoholic fatty liver disease.
Randomized controlled trial data
It is therefore hypothesized that a vegetarian diet would be superior to an omnivorous diet in reversing NAFLD and metabolic parameters [63]. This was not borne out, however, in a small randomized prospective study comparing the influence of a 6-week isocaloric high-protein diet using either plant or animal protein on liver fat and lipogenic indices in patients with T2DM and NAFLD. While the high animal protein diet led to large postprandial increases in BCAA and methionine compared to the plant protein group, both groups experienced significant improvements in FGF21, reductions in liver fat and down-regulation of lipolysis [64]. The results may have been similar due to small sample size and significant weight loss in both groups. Furthermore, the type of animal protein consumed (red, processed, lean) was not described.
Clinical advice
While large, prospective, RCTs are lacking to determine the impact of animal protein on the progression of NAFLD, it is reasonable to advise patients with NAFLD to reduce their intake of red and processed meats in light of their increased cardiovascular risk.
CHOS
Observational studies
Studies examining the association between CHOs and NAFLD are heterogeneous due to lack of differentiation between refined and unrefined CHOs. Low-diets have been associated with higher all-cause and cardiovascular mortality despite their benefits on initial weight loss, presumably due to reduced intake of unrefined CHOs (which are high in fiber, antioxidants, minerals, and vitamins), and increased consumption of animal protein, cholesterol and SFAs [65]. In a small study evaluating intestinal permeability, patients with NAFLD were found to have significantly higher intake of protein and CHOs, specifically mono- and disaccharides, compared to controls [66]. Protein and CHO intake correlated to higher alanine aminotransferase (ALT) levels in this group. High CHO intake (>70% of overall energy intake) has also been associated with higher aminotransferases and presence of the metabolic syndrome in a large Korean cohort, which persisted after adjustment for overall energy intake and BMI [67]. The opposite was found in a Portugal cohort comparing the diets of 45 patients with NASH to 856 controls, where lower CHO consumption was seen in patients with NASH, although this was accompanied by other differences in dietary composition [68]. A European cohort of 55 NAFLD patients and 88 controls found no significant differences in the relative intake of CHOs or fructose, although patients with NAFLD consumed more calories overall [16]. In children, total CHO intake has also been shown to be significantly higher in obese children with NAFLD compared to those without [69]. CHO intake has also been shown to increase in parallel to the degree of liver fat detected by ultrasonography [69].
No studies thus far directly compares the impact of refined versus unrefined CHOs on NAFLD, however there are several studies evaluating refined CHOs alone. In two small studies, consumption of dietary fructose has been shown to be significantly higher among NAFLD patients compared to controls [70,71]. SSBs, a surrogate for free sugars, have been associated with NAFLD in an Israeli population based study independent of age, gender, BMI and total caloric intake [44,72]. In the Framingham Heart Study cohort, higher consumption of SSBs incrementally increased the odds ratio for NAFLD across quartiles of consumption, even after adjustment for BMI, energy intake, dietary fiber, fat, protein and diet soda [72]. SSB consumption was also positively associated with ALT levels in this group. After controlling for dietary composition and physical activity, SSB consumption has been shown to be an independent variable to predict NAFLD with sensitivity of 100%, specificity 76%, positive predictive value 57% and negative predictive of 100% [73].
The data on histologic impact of CHO consumption is limited. When comparing nutrient intake of 28 patients with biopsy-proven NASH to 18 with simple steatosis, those with NASH had higher intake of CHOs, specifically simple CHOs [74]. In a bariatric surgery cohort, higher CHO intake was significantly associated with inflammation, but not fibrosis, on liver biopsy [7]. In older adults with NAFLD, higher daily fructose consumption has been associated with fibrosis, hepatic inflammation and hepatocyte ballooning [75].
Pathophysiology
CHOs induce DNL by activating the CHO responsive transcription factor, CHO response element binding protein (Fig. 1) [76]. Fructose is metabolized predominantly in the liver where it is converted into glyceraldehyde-3-phosphate, which can be used for gluconeogenesis or acetyl-CoA production [77]. The latter can be oxidized, or used for lipogenesis. Diets high in fructose contribute to NAFLD by increasing DNL and reducing fatty acid oxidation [78-81]. Fructose can also activate fatty acid synthase and stearoyl-CoA-desaturase-1 [82], sensitizing the liver towards inflammation, which may promote the development of NASH [83]. Although data are lacking, unrefined CHOs are likely to be protective against NAFLD as a result of their low glycemic index, high fiber content, and role in increasing production of short-chain fatty acids in the gut [84].
Randomized controlled trial data
Several RCTs have been performed to evaluate the effects of a low CHO diet (<50% of daily calories) on NAFLD. In attempt to consolidate the data, a meta-analysis of 10 RCTs was performed, however significant heterogeneity was encountered [85]. The overall conclusion was that a low CHO diet could reduce intrahepatic lipid content (IHLC) by over 10%, however significant weight loss across all intervention groups limited the ability to isolate the effects of a low CHO diet alone. Two of the trials evaluated a ketogenic diet (8% CHO in one, <20 g CHO per day in the other) over 2 and 24 weeks, respectively [86,87]. Despite weight loss in both, there was no significant reduction in ALT, however the 2 weeks study did show reduction in liver fat content. When low (<60 g/day) and high CHO (>180 g/day) diets were compared over an 11 weeks calorie restriction intervention, reduction in IHLC was comparable between both groups after 7% weight loss [24]. Furthermore, a direct comparison of hypocaloric diets (30% energy restricted) either low in CHO (and high in fat), or low in fat in 170 overweight individuals for 6 months, revealed comparable decreases in body weight, visceral fat and IHLC [88]. A meta-analysis of 13 trials with a total of 260 participants reported an association between a high fructose diet and NAFLD incidence and severity [89]. Only seven of these trials were isocaloric however, and in these studies fructose had no significant effect on IHLC or ALT [89]. Unfortunately the majority of studies were small, of short duration and marred by confounding factors. Similarly a subsequent meta-analysis of 21 interventional studies found only low levels of evidence that a high fructose diet was associated with increased liver fat content and transaminases as studies were significantly confounded by excess energy intake [90].
In adolescent boys, those on a limited free sugar diet (<5% daily calories) for 8 weeks experienced a significant decrease in hepatic steatosis and aminotransferases compared to those with no dietary intervention, though weight loss was greater in the intervention cohort [91]. Focusing specifically on fructose reduction (by 50%), a small pilot study showed reduction in IHLC and aminotransferases over 6 months [92]. Results are difficult to interpret given lack of control group as well as significant reduction in weight, SFA and sucrose intake. Over-feeding studies further highlight the association between SSB and NAFLD. When randomized to 1 L daily of sugar sweetened soda, skim milk, diet soda, or water for 6 months, those consuming the sugar sweetened soda had significant increases in IHLC, visceral fat, and skeletal muscle fat, compared to no changes in the other groups, despite no significant weight changes [93]. After overfeeding overweight adults with 1,000 kcal/day of candy or SSBs for 3 weeks, body weight increased by 2% accompanied by 27% increase in IHLC [94]. IHLC returned to baseline once baseline weight was achieved over the following 6 months. Whole, unrefined CHOs are protective against cardiovascular disease, T2DM, colorectal and breast cancer [95], and are associated with decreased all-cause mortality [96], however data in NAFLD is lacking.
Clinical advice
Clinicians should advise reduction in refined CHOs, specifically fructose, in patients with NAFLD.
FIBER
Observational studies
High fiber consumption is associated with a 15–30% decrease in all-cause and cardiovascular-related mortality, lower risk of heart disease, stroke, T2DM and gastro-intestinal cancer [95]. Epidemiological studies suggest there may be an association between a low fiber diet and the development of NAFLD. In a study of 45 patients with NASH and over 800 controls, more than onethird of patients with NASH consumed lower than the recommended requirements of fiber, although this did not reach statistical significance compared to the controls unless the consumption of soluble fiber was considered in isolation (in this case nearly 90% of patients consumed less than 10 g/day) [68]. Similarly, in an observational study of 55 NAFLD patients and 88 controls, NAFLD patients were found to consume less fiber [16]. These findings were supported by a case-control study from Iran (NAFLD, n=159; controls, n=158) [97], however both studies are limited by differing macronutrient composition compared to controls. Fiber consumption has also been shown to be significantly lower in obese children with moderate and severe hepatic steatosis, compared to obese children without NAFLD [69]. In a small uncontrolled pilot study, liver enzymes normalized in 75% of NAFLD patients eating 10 g/day of soluble fiber for 3 months [98]. This study is limited not only by lack of a control group, but also reduction in BMI, waist circumference, insulin resistance index and cholesterol levels in two thirds of patients.
Pathophysiology
The majority of studies looking at the benefits of fiber on cardiovascular risk factors have focused on soluble fiber. Soluble fiber is thought to be protective against NAFLD by reducing serum low-density lipoprotein (LDL)-cholesterol levels [99]. The mechanism for this is unclear, although it has been proposed that soluble fiber may bind to bile acids or cholesterol during the formation of micelles, lowering the cholesterol concentration in hepatocytes, leading to up-regulation of LDL receptors and clearance of LDL-cholesterol [100]. Soluble fiber can also slow the rate at which CHOs are absorbed into the circulation reducing post-prandial hyperglycemia, i.e., high fiber foods have a low glycemic index leading to improved glucose tolerance [101].
Randomized controlled trial data
RCTs studying the effect of fiber intake in isolation on NAFLD are lacking, as this usually forms part of a wider dietary intervention. In one study, 70 obese individuals with features of the metabolic syndrome were randomized to two energy-restricted diets for 6 months [102]. Participants who consumed higher levels of fiber from fruit experienced improvements in their FLI, hepatic steatosis index, NAFLD liver fat score and liver enzymes, supporting the consumption of fiber in the context of energy restriction for patients with NAFLD. In a small, randomized double-blind crossover trial, seven patients with NASH received 16 g/day of oligofructose (a prebiotic fiber), or placebo for 8 weeks. Oligofructose led to a significant reduction in insulin levels, as well as ALT and aspartate aminotransferase after 4 and 8 weeks, respectively, independent of a significant effect on plasma lipids [103].
Clinical advice
Increasing fiber intake by increasing intake of fruits, vegetables, whole grains and legumes, should be encouraged in patients with NAFLD.
FATS
Observational studies
There is a strong degree of concordance between observational studies to show that a higher intake of SFAs [68,69,74,97,104], and a lower intake of polyunsaturated fatty acids (PUFAs) [69,74,104], is associated with NAFLD and NASH. In a cohort of 25 patients with NASH and 25 BMI matched controls, 7-day alimentary records revealed that patients with NASH consumed significantly higher proportions of SFAs and a lower percentage of PUFAs, although differences were also seen between their intake of cholesterol, fiber and anti-oxidant vitamins [104]. The ratio of PUFAs to SFAs was also lower in patients with NASH and NAFLD compared to the general population in a Japanese cohort [74]. These findings were replicated in pediatric cohort in which SFA intake correlated proportionally to the degree of hepatic steatosis [69]. Furthermore, omega-3 fatty acid consumption was lower in pediatric NAFLD patients and this, along with insulin resistance, remained the most significant factor following multiple regression analysis. Cortez-Pinto et al. [68] also reported higher total fat consumption in NASH patients compared to controls, including higher consumption of omega-6 fatty acids, which differs from other studies. This study may have been confounded by differences in CHO and fiber consumption between groups.
Pathophysiology
SFAs exert their effects on the liver through the promotion of insulin resistance and oxidative stress. They induce hepatic steatosis by increasing lipolysis as well as DNL, which occurs through the promotion of the transcription of peroxisome proliferator-activated receptor (PPAR) γ coactivator-1β and SREBP-1c (Fig. 3) [105]. SFAs also promote lipotoxicity through ceramides and diacylglycerides [106], and can induce hepatocyte apoptosis and increase oxidative stress, which may encourage progression towards NASH [107]. Conversely, monounsaturated fatty acids (MUFAs) activate transcription factors PPARγ and PPARα, promoting safe fatty acid storage in adipose tissue and lipid detoxification via fatty acid oxidation, respectively [108]. PUFAs increase the transcription of PPARα, increasing lipid metabolism and mitochondrial oxidation, thereby reducing hepatic FFA concentrations [109,110]. They also inhibit SREBP-1c, reducing fatty acid synthesis [111,112]. Omega-3 fatty acids lower the hepatic triglyceride content by suppressing hepatic VLDL apolipoprotein B-100 [113], and inhibit inflammatory cells involved in NASH [114].

Mechanisms via which saturated and unsaturated fatty acids influence the pathogenesis of NAFLD. PPAR, peroxisome proliferator-activated receptor; FFAs, free fatty acids; VLDL, very low density lipoprotein; SREBP-1c, sterol regulatory element-binding protein-1c; NAFLD, nonalcoholic fatty liver disease.
Randomized controlled trial data
The beneficial effects of PUFAs and MUFAs, and metabolically harmful effects of SFAs, are shown in Table 2 [115-119]. Two meta-analyses have examined the effects of omega-3 supplementation on NAFLD [120,121]. The number of eligible studies analysed were nine and 10, respectively (335 individuals and 577 individuals with NAFLD). Both reported that omega-3 supplementation was beneficial in reducing liver fat (predominantly quantified using ultrasound), but did not impact liver biochemistry. The analyses, however, were limited by poor quality study design and heterogeneity. It was not possible to comment on an optimal dose. In terms of histology, a trial comparing the effect of a diet high in PUFAs vs. placebo for 1 year in individuals with NASH revealed no significant difference in NAS (≥2 point reduction) despite a greater reduction in liver fat in the treatment group, although participant numbers were small (n=34) [122].
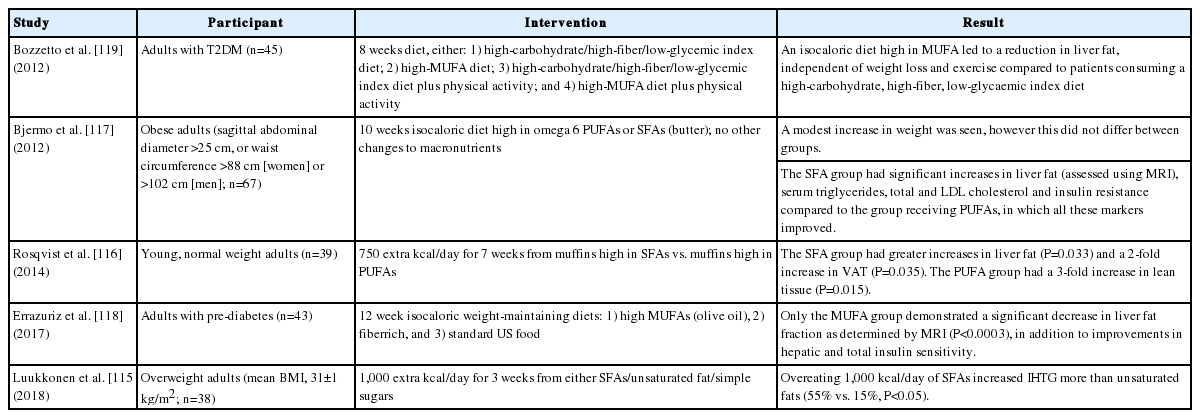
Summary of randomized controlled trial data examining the influence of diets high in saturated fatty acids and poly- and mono-unsaturated fatty acids on hepatic steatosis
Clinical advice
Clinicians should advise patients with NAFLD to replace dietary SFAs with PUFAs or MUFAs.
DIETS
The Dietary Approaches to Stop Hypertension (DASH) diet is a low-glycemic, low energy-dense diet characterized by high intake of fruits and vegetables, whole grains, and low fat dairy products, with limited SFAs. An RCT comparing patients with NAFLD eating the DASH diet versus a control diet for 8 weeks (both of which contained 52–55% CHOs, 16–18% protein, 30% fat and approximately 1,900 kcal/day), showed that the DASH group had significantly greater reduction in aminotransferases and metabolic markers, including serum triglycerides, total cholesterol, VLDL cholesterol, high sensitivity c-reactive protein, insulin and Homeostatic Model Assessment of Insulin Resistance (HOMA-IR) [123]. The data are confounded by greater weight loss in patients following the DASH diet, however the high fiber and antioxidant content, and low saturated fat and refined CHOs content, is likely to be beneficial for NAFLD.
Soy is also thought to be helpful in NAFLD by inhibiting SREBP-1c and activating PPARα, reducing lipid deposition and increasing antioxidant capacity. A three-arm RCT comparing patients eating a low calorie diet to a low-CHO, low calorie diet to a low-CHO, low calorie, soy-containing diet, reported that individuals eating the soy-containing diet had significantly greater improvements in liver tests and serum insulin levels [124].
While RCT data is sparse, research suggests that the Mediterranean diet (rich in plant-based foods, legumes and unsaturated fats) should prove ideal for patients with NAFLD as a result of its effectiveness as a form of primary prevention for components of the metabolic syndrome, and ability to reduce insulin resistance, liver fat and inflammation [125]. An ad libitum Mediterranean diet compared to a low-fat diet showed similar reductions in hepatic steatosis over 12 weeks with similar weight loss in both groups. The Mediterranean group alone, however, saw improvements in cholesterol, triglycerides and hemoglobin A1C [126]. Furthermore, obese individuals with diabetes asked to follow a modified Mediterranean diet for 12 months were found to display lower levels of ALT compared to participants allocated to the American Diabetes Association diet and a low glycaemic index diet [127]. The Mediterranean diet is now recommended by EASL for the management of NAFLD [14].
Clinical advice
For patients with NAFLD we recommend diets high in whole, unprocessed foods, fiber, and unsaturated fats, with limited quantities of red and processed meats, refined CHOs and saturated fat. Example diets include the Mediterranean diet, DASH diet, and other plant-based diets.
BEVERAGES
Two large systematic reviews have shown that coffee leads to a relative risk reduction of cirrhosis and liver-related mortality secondary to all causes [128,129]. In terms of NAFLD, two meta-analysis have demonstrated that coffee can reduce the incidence of NAFLD, in addition to decreasing the risk of liver fibrosis among patients with established NAFLD [130,131]. A non-linearity curve relationship between coffee consumption and the development of NAFLD is described, with more than 3 cups per day reducing the incidence of NAFLD significantly [130]. Several constituents found within coffee have been postulated as being mechanistic due to their favourable effects on glucose metabolism [132]. For example, chlorogenic acid inhibits glucose-6-phosphate hydrolysis, leading to a reduction in gluconeogenesis and glycogenolysis, and can inhibit glucose absorption from the gut [133].
Concomitant alcohol consumption is frequently encountered in patients with NAFLD. Previous meta-analyses have shown no association between alcohol intake (up to 80 g/day) and hepatic steatosis [134], and a protective effect for individuals drinking up to 40 g/day [135], however these studies were largely heterogenous, retrospective and subject to selection bias. A cross-sectional study concluded that steatosis is present in nearly 95% of obese persons who drink more than 60 g of alcohol per day, however obesity plays the over-arching role [136]. There is, however, strong evidence that patients drinking excessively (≥2 drinks/day for women and ≥3 drinks/day for men) with NAFLD are at significantly increased risk of developing advanced liver fibrosis and this should therefore be discouraged [137]. Even mild to moderate drinking (<210 g/week) has been found to increase the risk of steatohepatitis, fibrosis, decompensated liver disease, mortality and liver cancer among individuals with obesity and diabetes [138-144], although there is some disagreement among studies [135,145,146]. Abstinence has been advocated for patients with NASH cirrhosis in order to reduce the risk for decompensation and hepatocellular carcinoma (HCC) [147].
Clinical advice
Coffee consumption is protective against the development of NAFLD and disease progression. Moderate to heavy alcohol consumption should be avoided in the presence of obesity, NAFLD, and other metabolic risk factors. Abstinence is advised for patients with advanced fibrosis.
LIMITATIONS OF CURRENT DATA
One of the biggest challenges encountered when studying dietary determinants of diseases is the confounding effects of other dietary components and lifestyle factors. This may not be easily handled by multivariable analyses alone and can lead to erroneous conclusions. These issues were recently demonstrated in a series of meta-analyses which concluded that red or processed meat may only lead to small differences in the risk of all-cause mortality, cardiometabolic outcomes and cancer incidence [148-151]; in contrast to the established medical opinion and public beliefs. Three of the reviews were reliant on observational studies for which many received low GRADE scores in terms of evidence quality. The authors identified fundamental issues inherent to the design of many nutritional studies including a lack of a clear hypothesis, selective reporting of results, reliance on self-reported food consumption, lack of controls and failure to address confounders [152,153]. A review of RCT data comparing diets with differing amounts of red meat consumed for at least 6 months was similarly afflicted by poor quality evidence and discordant results [150]. These results were used to inform guidelines published by the Nutritional Recommendations (NutriRECS) Consortium, which are the first to suggest there is no need for adults to reduce their consumption of red and processed meat [154], and have received widespread public criticism. It is vital that we are able to provide the public with accurate information about their dietary choices and maintain the integrity of evidence-based medicine. There therefore needs to be radical reform in terms of how these studies are undertaken. Small, poorly design observational studies are perhaps damaging to these field; instead investment is required in high quality large RCTs looking at long term outcomes, in addition to the collection of longitudinal data on markers of early disease. We have summarized these research priorities in Table 3.

Suggested research priorities
CONCLUSION
This review has highlighted a significant lack of high quality RCT data in this field, offering a number of research opportunities for the future. Although well intentioned, diets focusing specifically on reducing CHO or fat intake miss out on the benefits of whole grains, fiber, and unsaturated fats, which do not need to be minimized in the diet. These diets are also not sustainable. Instead, the focus should shift to a lifestyle that incorporates healthy CHOs and fats, which may be more sustainable long term. Overall the current data is supportive of diets low in SFAs, red and processed meats, and refined CHOs for NAFLD. Diets that incorporate these recommendations include plant-based diets such as the DASH, Mediterranean, vegetarian, and vegan diets.
Notes
Authors’ contributions
Theresa J. Hydes: Drafting the article, critical revision of the article and final approval of the version to be published
Sujan Ravi: Drafting the article, critical revision of the article and final approval of the version to be published
Rohit Loomba: Critical revision of the article and final approval of the version to be published
Meagan E. Gray: Conception and design, drafting the article, critical revision of the article and final approval of the version to be published
Conflicts of Interest
The authors have no conflicts to disclose.
Acknowledgements
No grant or financial support of any of the authors is related to this work. MG receives funding support from NIDDK (R01DK108353). RL receives funding support from NIEHS (5P42ES010337), NCATS (5UL1TR001442), NIDDK (R01DK106419, P30DK120515), and DOD PRCRP (CA170674P2). Potential competing interests: MG serves as an advisory board member for Intercept. RL serves as a consultant or advisory board member for Arrowhead Pharmaceuticals, AstraZeneca, Bird Rock Bio, Boehringer Ingelheim, BristolMyer Squibb, Celgene, Cirius, CohBar, Conatus, Eli Lilly, Galmed, Gemphire, Gilead, Glympse bio, GNI, GRI Bio, Intercept, Ionis, Janssen Inc., Merck, Metacrine, Inc., NGM Biopharmaceuticals, Novartis, Novo Nordisk, Pfizer, Prometheus, Sanofi, Siemens, and Viking Therapeutics. In addition, his institution has received grant support from Allergan, Boehringer-Ingelheim, Bristol-Myers Squibb, Cirius, Eli Lilly and Company, Galectin Therapeutics, Galmed Pharmaceuticals, GE, Genfit, Gilead, Intercept, Grail, Janssen, Madrigal Pharmaceuticals, Merck, NGM Biopharmaceuticals, NuSirt, Pfizer, pH Pharma, Prometheus, and Siemens. He is also co-founder of Liponexus, Inc.
Abbreviations
ALT
alanine aminotransferase
BCAA
branched chain amino acids
BMI
body mass index
CHO
carbohydrates
DASH
Dietary Approaches to Stop Hypertension
DNL
de novo lipogenesis
EASL
European Association for the Study of the Liver
FFAs
free fatty acids
FGF21
fibroblast growth factor 21
FLI
fatty liver index; HCC, hepatocellular carcinoma
HOMA-IR
Homeostatic Model Assessment of Insulin Resistance
IF
intermittent fasting
IHLC
intrahepatic lipid content
LDL
low-density lipoprotein
mTOR
mammalian target of rapamycin
MUFA
monounsaturated fatty acids
NAFLD
nonalcoholic fatty liver disease
NAS
NAFLD activity score
NASH
nonalcoholic steatohepatitis
NutriRECS
Nutritional Recommendations
PPAR
peroxisome proliferator-activated receptor
PUFAs
polyunsaturated fatty acids
RCT
randomized controlled trial
SFAs
saturated fatty acids
SREBP-1c
sterol regulatory element-binding protein-1c
SSBs
sugar-sweetened beverages
T2DM
type 2 diabetes mellitus
TMA
trimethylamine
TMAO
trimethylamine N-oxide
US
United States
VAT
visceral adipose tissue
VLDL
very low-density lipoprotein