Obesity and binge alcohol intake are deadly combination to induce steatohepatitis: A model of high-fat diet and binge ethanol intake
Article information

Abstract
Obesity and binge drinking often coexist and work synergistically to promote steatohepatitis; however, the underlying mechanisms remain obscure. In this mini-review, we briefly summarize clinical evidence of the synergistical effect of obesity and heavy drinking on steatohepatitis and discuss the underlying mechanisms obtained from the study of several mouse models. High-fat diet (HFD) feeding and binge ethanol synergistically induced steatohepatitis and fibrosis in mice with significant intrahepatic neutrophil infiltration; such HFD-plus-ethanol treatment markedly up-regulated the hepatic expression of many chemokines with the highest fold (approximately 30-fold) induction of chemokine (C-X-C motif) ligand 1 (Cxcl1), which contributes to hepatic neutrophil infiltration and liver injury. Furthermore, HFD feeding activated peroxisome proliferator-activated receptor gamma that subsequently inhibited CXCL1 upregulation in hepatocytes, thereby forming a negative feedback loop to prevent neutrophil overaction; whereas binge ethanol blocked this loop and then exacerbated CXCL1 elevation, neutrophil infiltration, and liver injury. Interestingly, inflamed mouse hepatocytes attracted neutrophils less effectively than inflamed human hepatocytes due to the lower induction of CXCL1 and the lack of the interleukin (IL)-8 gene in the mouse genome, which may be one of the reasons for difficulty in development of mouse models of alcoholic steatohepatitis and nonalcoholic steatohepatitis (NASH). Hepatic overexpression of Cxcl1 and/or IL-8 promoted steatosis-to-NASH progression in HFD-fed mice by inducing neutrophil infiltration, oxidative stress, hepatocyte death, fibrosis, and p38 mitogen-activated protein kinase activation. Collectively, obesity and binge drinking synergistically promote steatohepatitis via the induction of CXCL1 and subsequent hepatic neutrophil infiltration.
INTRODUCTION
Although obesity and heavy drinking, which often coexist in patients, have been shown to promote the development of liver injury, steatohepatitis, and fibrosis [1,2], the clinical data on light-tomoderate alcohol drinking on nonalcoholic fatty liver disease (NAFLD) have been controversial [3,4]. The data from experimental models on the effect of chronic alcohol feeding on high-fat diet (HFD)-induced NAFLD are limited because it is difficult to manage chronic alcohol and HFD feeding together due to marked reduction of ad libitum HFD intake by chronic ethanol feeding in mice. Xu et al. [5] performed intragastric force-feeding of HFD-plus-ethanol in mice and found HFD and ethanol synergistically induce nitrosative, endoplasmic reticulum (ER), and mitochondrial stress and an upregulation of hepatic toll-like receptor 4, thereby contributing to steatohepatitis. Recently, we developed a model of HFD-plus-binge ethanol challenge [6,7], which mimics binge drinking and obesity in humans. Our data revealed that binge alcohol intake and HFD synergistically induce steatohepatitis and fibrosis [6,7]. In the current review, we briefly describe clinical data on the effects of obesity and alcohol drinking on steatohepatitis and deliberate the underlying mechanisms obtained from the study of several mouse models.
CLINICAL DATA ON THE EFFECTS OF OBESITY AND ALCOHOL DRINKING ON NAFLD
Recent increases in the incidence of obesity and metabolic syndrome have highlighted the potential synergism between obesity and heavy drinking to cause liver injury and resultant inflammation [1,2]. Several clinical studies have documented the additive effects of obesity and heavy/moderate alcohol drinking on steatohepatitis and liver-related death [8-13]. For example, in a cohort study of 233 alcoholic hepatitis patients, obesity was found to be associated with a greater than two-fold increase in short-term mortality [2]. However, the clinical data on the effects of light to moderate alcohol drinking on NAFLD have been controversial. Some early observational studies suggest low to moderate alcohol use does not increase or even reduces the risk of fatty liver [14-16]; but this conclusion was later questioned by meta analyses [4,17]. The cross-sectional profile of these studies make it impossible to establish temporal relationships between alcohol consumption and NAFLD. A Korean cohort study followed 190,048 adults without NAFLD at baseline for 15.7 years and documented a decreased risk of hepatic steatosis in moderate drinkers [18]. However, in the subgroup of participants who developed more severe NAFLD over time, low levels of alcohol consumption were associated with the risk of liver fibrosis, especially for nonobese individuals [18]. Besides the impact on hepatic steatosis incidence, the impact of low to moderate drinking on NAFLD development and relevant clinical outcomes has received fierce debate as well. Targeting biopsyconfirmed NAFLD patients, moderate drinking was reported to bring about lower odds for histological improvement and nonalcoholic steatohepatitis (NASH) resolution [19]. In a large-scale Korean cohort of young and middle-aged NAFLD individuals, nonheavy alcohol use was associated with noninvasive fibrosis indexes, indicating the perniciousness of even modest drinking [10]. Longitudinal observation on more severe NAFLD cases with fibrosis or cirrhosis further demonstrated the facilitation of low to moderate drinking to liver decompensation and liver-related death [20]. A recent study on Finland population also showed a correlation between alcohol intake (even at low levels) and the risk for advanced liver diseases on the basis of fatty liver. But as a whole, low to moderate drinking was associated with reduced mortality and cardiovascular disease (CVD) risk in NAFLD patients [8]. Similarly, a prospective study based on 4,568 USA subjects with NAFLD (defined by the hepatic steatosis index) noted significant protective effects of 0.5–1.5 drinks per day against allcause mortality, whereas ≥1.5 drinks per day led to an increase in mortality [21]. In contrast, a longitudinal cohort study following 570 USA individuals with NAFLD found prospectively assessed alcohol use was not associated with significant differences in CVD risk [22]. These results were further challenged by the Global Burden of Disease study, which indicated even light drinking increases CVD risk and mortality [23]. Given such controversy and disagreement regarding the effects of alcohol on NAFLD, further rigor clinical and mechanistic studies are in urgent need. Table 1 summarized recent clinical studies on liver disease caused by combination of alcohol drinking and obesity.

Recent clinical studies on liver disease caused by combination of ethanol and obesity
HFD-PLUS-ETHANOL BINGE MODEL INDUCES SEVERE STEATOHEPATITIS
Investigators have recently performed experimental studies to better understand the synergistic role of ethanol and obesity and the associated mechanisms for the potentiation of liver disease by ethanol and obesity as summarized in Table 2. Because chronic alcohol feeding markedly reduces ad libitum HFD intake in mice, it is difficult to manage chronic alcohol and HFD feeding together unless performing intragastric force-feeding [5]. Recently, we developed a model to study the interaction of obesity and binge drinking by introducing binge alcohol intake in HFD-fed mice, and our data suggest that binge ethanol intake and HFD synergistically induced steatohepatitis as shown by a marked increase in hepatic neutrophil infiltration and inflammatory markers as well as liver injury (Fig. 1) [6].
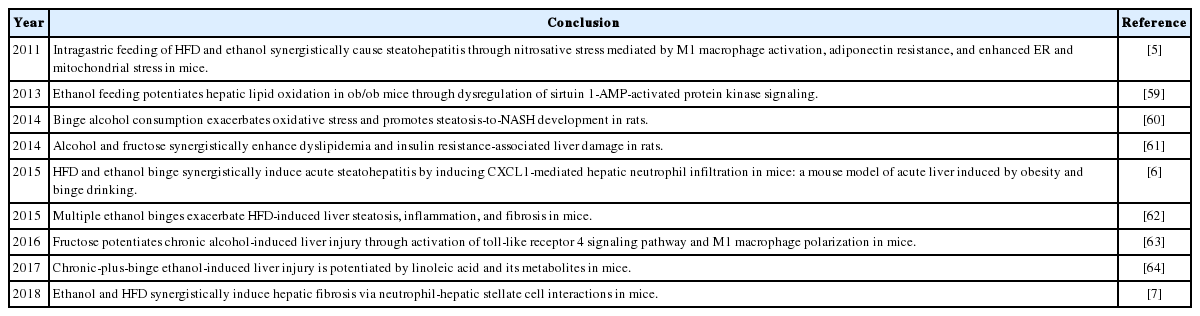
Recent experimental studies on liver disease caused by combination of ethanol and obesity
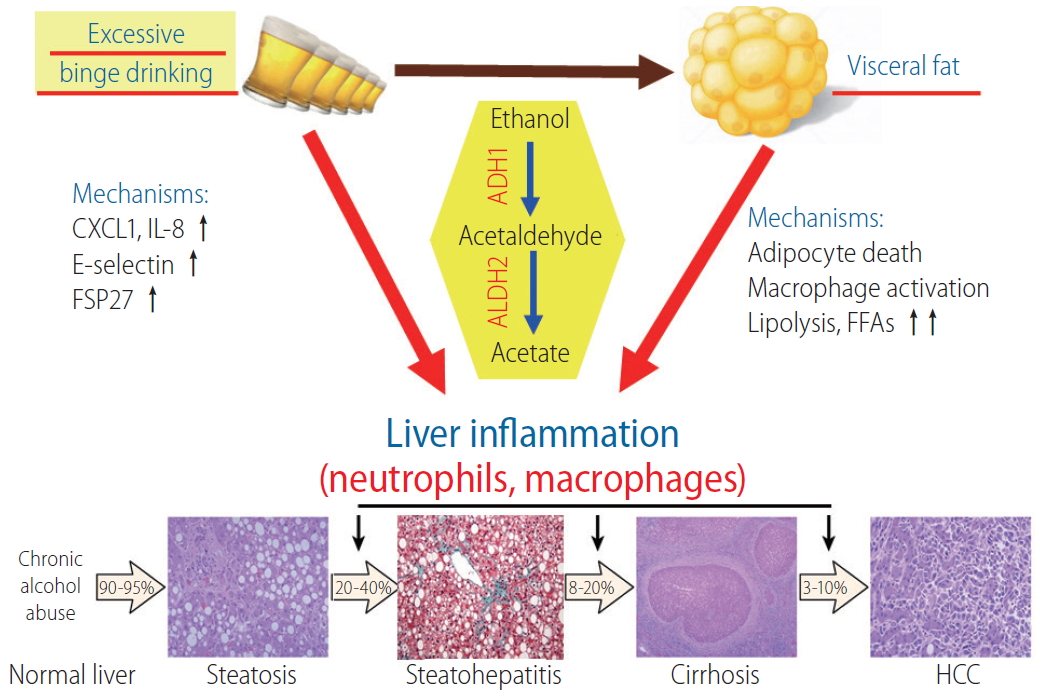
Binge drinking and fat synergistically promote ALD. Ingested ethanol is metabolized to acetaldehyde and acetate by the sequential action of ADH1 and ALDH2, respectively. Excessive binge drinking induces neutrophil-recruiting chemokines such as CXCL1 and IL-8 and endothelial cell adhesion molecule E-selectin, thereby allowing for the enhanced infiltration of neutrophils in the liver. Binge drinking also results in mitochondrial DNA damage and induction of fat-accumulating protein FSP27 in the liver. In cooperation with these factors that enhance liver injury and inflammation, alcohol-induced dysregulation of visceral fat potentiates the liver inflammation through mechanisms involving adipocyte death, macrophage activation, lipolysis, and FFA release. IL-8, interleukin 8; FSP27, fat-specific protein 27; ADH1, alcohol dehydrogenase 1; ALDH2, aldehyde dehydrogenase 2; FFA, free fatty acid; HCC, hepatocellular carcinoma; ALD, alcohol-related liver disease; CXCL1, C-X-C motif chemokine ligand 1.
Hepatic neutrophil infiltration is a critical pathologic feature during liver injury, usually serving as a hallmark of both obesity and alcohol-related liver diseases [24]. C-X-C motif chemokine ligand 1 (CXCL1) belongs to the CXC chemokine family and is the major chemokine mediating neutrophil recruitment through CXCR2 [25]. In patients with alcoholic hepatitis, both hepatic and serum levels of CXCL1 were reported markedly elevated. Meanwhile, hepatic Cxcl1 mRNA levels correlated positively with hepatic neutrophil infiltration in the same group of patients [26]. More remarkably, the Cxcl1 rs4074 A allele in human has been associated with elevated serum CXCL1 levels and considered as a genetic risk factor for alcoholic cirrhosis [27]. Cxcl1 was one of the most highly-induced genes in the liver of mice upon challenge with the combination of 3-month HFD-plus-binge ethanol (30-fold in the liver and 5-fold in epididymal adipose tissue) [6]. In the liver, elevation of Cxcl1 expression was the most significant in hepatocytes, and to a lesser extent in hepatic stellate cells (HSCs) and liver sinusoidal endothelial cells [6]. The induction of CXCL1 by HFD-plus-ethanol binge is believed to be associated with the increased levels of free fatty acid (FFA) in the liver, which upregulate Cxcl1 in hepatocytes in a manner depending on extracellular signal-regulated kinase 1/2, c-Jun N-terminal kinase (JNK), and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling. Blockade of CXCL1 with a neutralizing antibody or disrupting the Cxcl1 gene ameliorated hepatic neutrophil infiltration and injury after HFD-plus-ethanol binge, whereas overexpression of Cxcl1 exacerbated steatohepatitis in 3-motnh HFD-fed mice.
Peroxisome proliferator-activated receptor gamma (PPAR-γ) expression in the liver is very low under physiological conditions, but markedly upregulated in fatty liver animal models and NAFLD patients [28]. Disruption of the Pparg gene in hepatocytes ameliorates steatosis, whereas its overexpression promotes the development of fatty liver through the activation of various lipogenic genes, including fat-specific protein 27 (FSP27) [29], and the resultant lipogenesis [30]. The steatohepatitis model featuring the combination of HFD-plus-ethanol binge identified the dichotomous role of PPAR-γ underlying the synergistic effect of HFD and ethanol binge on steatohepatitis: 1) HFD feeding activates PPAR-γ, and FFA upregulates hepatic CXCL1 expression; 2) PPAR-γ inhibits NF-κB-mediated Cxcl1 expression and subsequently prevents hepatic neutrophil infiltration; and 3) ethanol binge blocks this negative feedback loop and increases the NF-κB/CXCL1 pathway, which consequently exacerbates neutrophil infiltration and liver injury. In addition, this HFD-plus-binge ethanol model has been recently used to demonstrate that γδT cells play a central role in steatohepatitis by mitigating T cell expansion and modulating their inflammatory program [31,32]. This model was also recently used to test some therapies for alcohol-induced liver toxicity [33].
Although this HFD-plus-ethanol binge model does not represent chronic alcoholic steatohepatitis (ASH), it can trigger acute liver injury and mimic hepatic neutrophil infiltration as typically shown in ASH patients [34,35]. Its obvious clinical implication is that obese individuals should avoid binge drinking of large amount of alcohol, as this would likely cause significant acute liver damage. Furthermore, the rapidity of the development of neutrophil-mediated steatohepatitis, which would make it uniquely suitable for future mechanistic studies.
HFD-PLUS-ETHANOL BINGE PROMOTES LIVER FIBROSIS
During steatohepatitis, persistent inflammation dysregulates liver homeostasis and activates the repair processes [36]. Activated HSCs secrete extracellular matrix to support the injured region and construct the regeneration scaffold for subsequent hepatocyte proliferation (i.e., liver fibrosis formation), along with the clearance of necrotic tissues by infiltrating leukocytes [37]. Fibrosis has been postulated to be promoted by neutrophils that induce hepatocellular damage and HSC activation via the production of reactive oxygen species (ROS) [36]. HFD-plus-ethanol binge that features the hepatic infiltration of neutrophils caused a marked upregulation of an array of genes related to HSC activation and fibrogenesis compared with HFD feeding only [7]. Blockade of CXCL1 with a neutralizing antibody or genetic disruption of the Cxcl1 gene markedly attenuated liver fibrosis in the HFD-plus-ethanol binge model, and disruption of the intercellular adhesion molecule 1 (ICAM-1) gene that encodes ICAM-1 protein, a key adhesion molecule for neutrophil recruitment, attenuated neutrophil infiltration and liver fibrosis caused by HFD-plus- binge ethanol, indicating the significant involvement of infiltrated neutrophils in mediating fibrogenesis [7]. Mechanistically, ROS production mediated by p47phox, an important component of nicotinamide adenine dinucleotide phosphate oxidase 2 complex that mediates neutrophil oxidative burst [38], potentiates HSC activation, and activated HSCs act against neutrophils death via the strong production of granulocyte-macrophage colony-stimulating factor and interleukin (IL)-15, which are growth factors that play crucial roles in sustaining the survival of neutrophils [39,40]. This result indicates that binge drinking potentiates fibrogenesis in mice fed an HFD [41], expanding on the synergism between obesity and binge drinking in exacerbation of liver injury and neutrophil infiltration.
ADIPOSE TISSUE DYSFUNCTION CONTRIBUTES TO ALCOHOL-INDUCED LIVER INJURY
In addition to the synergism between hepatic fat and alcohol toxicity in the liver, the crosstalk between the adipose tissue and the liver has been highlighted as the cause for worsening alcohol-induced liver injury in obese individuals [1]. It has been recently reported that alcohol consumption impairs the function of adipose tissue through induction of adipocyte apoptosis and subsequent inflammation as well as lipolysis of adipocytes, reduction in insulin-dependent glucose uptake, and dysregulation of adipokines and cytokines (Fig. 2) [42,43]. A study utilizing a mouse model of inducible adipocyte death demonstrated that acute adipocyte death results in liver injury and inflammation via lipolysis and the activation of chemokine (C-C motif) receptor 2-positive macrophages [44], supporting the importance of adipocyte dysfunction in alcohol-induced liver injury.
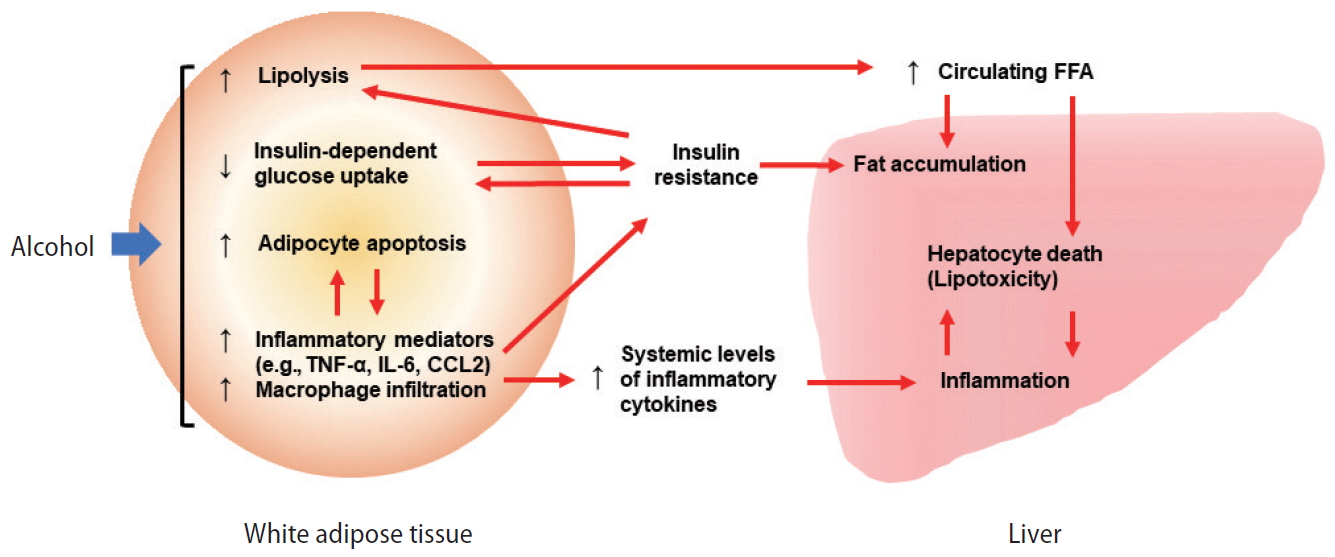
Role of white adipose tissue in the pathogenesis of ALD. Excessive alcohol intake induces white adipocyte death and white adipose tissue inflammation, resulting in elevation of epinephrine and norepinephrine, and subsequent lipolysis. Alcohol and dysfunctional white adipocytes can trigger insulin resistance, which also promotes lipolysis. White adipose tissue lipolysis causes elevation of circulating FFA levels, thereby inducing hepatic FFA influx, lipotoxicity, steatosis, hepatocyte death, and liver inflammation. Modified from Hwang and Gao [42]. TNF-α, tumor necrosis factor-alpha; IL-6, interleukin-6; CCL2, chemokine (C-C) motif ligand 2; FFA, free fatty acid; ALD, alcohol-related liver disease.
OVEREXPRESSION OF CXCL1 PROMOTES STEATOSIS-TO-NASH PROGRESSION
Obesity and insulin resistance are major risk factors for NAFLD, which ranges from steatosis to NASH, cirrhosis, and hepatocellular carcinoma [45,46]. Steatosis features excessive accumulation of fat in the liver without signs of severe inflammation or liver injury [47]. Approximately 25% of patients with steatosis further progress to NASH, which is defined by the presence of liver injury (hepatocyte ballooning), inflammation, and fibrosis in addition to hepatic fat deposition [47,48]. Diet-induced obesity model such as 3-month HFD feeding in mice readily results in the development of steatosis with gene expression profile and histological features similar to those of human steatosis; however, mice are relatively resistant to NASH development under this condition.
A key feature of human NASH is the marked infiltration of neutrophils in the liver and enhanced hepatic expression of CXCL1 and IL-8, major chemokines for neutrophil chemotaxis [49], which are not commonly observed in patients with fatty liver or mice subjected to HFD-induced fatty liver model. However, it has been elusive whether neutrophils play a critical role in steatosis-to-NASH progression, and if so, how infiltrated neutrophils contribute to the development of NASH.
Adenovirus-mediated overexpression of Cxcl1 significantly elevated neutrophil infiltration in the liver of mice fed an HFD for 3 months [50]. Increased neutrophil population promoted ROS production which led to an activation of apoptosis signal-regulating kinase 1 (ASK1) and p38 mitogen-activated protein kinase (MAPK), thereby activating the signaling pathways leading to apoptosis- and ER stress-induced hepatocyte death [51]. DNA damage, oxidative stress, and other intrinsic apoptotic stimuli incurred by infiltrated neutrophils activate the mitochondrial apoptotic pathway of hepatocytes. ROS production can also disturb the protein folding process within the ER lumen by changing the redox status, which further cause ER stress [52,53]. It has been also demonstrated that infiltrated neutrophils cause hepatocyte death through p47phox-dependent oxidative burst and subsequent activation of ROS-sensitive stress kinases such as p38 MAPK and JNK [50], which in turn facilitates inflammatory and fibrogenic processes and exacerbates steatosis-to-NASH progression. Microarray analysis revealed that HFD/CXCL1-induced NASH possessed expression profiles of inflammatory and fibrogenic genes, which are similar to those of NASH patients, supporting the notion that Cxcl1 overexpression results in steatosis-to-NASH progression in HFD-fed mice [50].
Experimental models widely used to study NASH are not developed based on the molecular mechanisms that promote steatosis-to-NASH progression, but mostly developed by modulating the components of diet as seen in high-fat high-cholesterol diet, methionine choline-deficient diet, and HFD-plus-chronic CCl4 treatment [54,55]. Hepatic neutrophil infiltration is commonly observed in steatohepatitis with different etiologies including alcohol consumption and metabolic dysregulation, and thus experimental model exploiting this hallmark of steatohepatitis will better recapitulate the spectrum of NAFLD progression in human and serve as a useful tool to investigate the pathogenic mechanisms of NASH as well as a novel therapeutic target.
CONCLUSION
Emerging data from experimental models clearly suggest that binge ethanol intake and obesity synergistically induce steatohepatitis in mice via the induction of hepatic CXCL1 and neutrophil infiltration. So far, the effects of binge drinking and obesity on steatohepatitis in humans have not been carefully studied. Interestingly, we recently demonstrated that inflamed human hepatocytes attracted neutrophils more effectively than inflamed mouse hepatocytes due to the greater induction of CXCL1 and IL-8 in human hepatocytes (lack of the IL-8 gene in the mouse genome) [51]. In addition, the number of circulating neutrophils in humans (~4×109/L) is much higher than in mice (~1×109/L) [56]. Finally, ethanol metabolism in humans is five times slower than that in rodents [57]. Based on these factors, it is plausible to speculate that the synergistic effect of binge drinking and obesity on steatohepatitis in humans may be more significant than that in mice. Thus, obese individuals should avoid binge drinking large amount of alcohol as this would likely cause significant acute liver damage.
Notes
Authors’ contribution
SH and TR wrote the paper, BG supervised the whole project and edited the paper.
Conflicts of Interest: The authors have no conflicts to disclose.
Acknowledgements
The work from Dr. Bin Gao’s lab described in the current paper was supported by the intramural program of NIAAA, NIH (B.G.).
Abbreviations
CVD
cardiovascular disease
CXCL1
C-X-C motif chemokine ligand 1
ER
endoplasmic reticulum
FFA
free fatty acid
FSP27
fat-specific protein 27
HFD
high-fat diet
HSCs
hepatic stellate cells
ICAM-1
intercellular adhesion molecule 1
IL
interleukin
JNK
c-Jun N-terminal kinase
MAPK
mitogen-activated protein kinase
NAFLD
nonalcoholic fatty liver disease
NASH
nonalcoholic steatohepatitis
ASH
alcoholic steatohepatitis
NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
PPAR-g
peroxisome proliferator-activated receptor gamma
ROS
reactive oxygen species
ASK1
apoptosis signal-regulating kinase 1