Nonalcoholic fatty liver disease and alcohol-related liver disease: From clinical aspects to pathophysiological insights
Article information

Abstract
Two major causes of steatohepatitis are alcohol and metabolic syndrome. Although the underlying causes of alcohol-related liver disease (ALD) and nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH) differ, there are certain similarities in terms of the mode of disease progression and underlying pathophysiological mechanisms. Further, excessive alcohol consumption is often seen in patients with metabolic syndrome, and alcoholic hepatitis exacerbation by comorbidity with metabolic syndrome is an emerging clinical problem. There are certain ethnic differences in the development of both NAFLD and ALD. Especially, Asian populations tend to be more susceptible to NAFLD, and genetic polymorphisms in patatin-like phospholipase domain-containing 3 (PNPLA3) play a key role in both NAFLD and ALD. From the viewpoint of pathophysiology, cellular stress responses, including autophagy and endoplasmic reticulum (ER) stress, are involved in the development of cellular injury in steatohepatitis. Further, gut-derived bacterial products and innate immune responses in the liver most likely play a profound role in the pathogenesis of both ALD and NASH. Though the recent progress in the treatment of viral hepatitis has reduced the prevalence of viral-related development of hepatocellular carcinoma (HCC), non-viral HCC is increasing. Alcohol and metabolic syndrome synergistically exacerbate progression of steatohepatitis, resulting in carcinogenesis. The gut-liver axis is a potential therapeutic and prophylactic target for steatohepatitis and subsequent carcinogenesis.
INTRODUCTION
Nonalcoholic fatty liver disease (NAFLD) is a clinical entity comprising a wide spectrum of liver diseases from nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH), a progressive form of chronic liver disease eventually resulting in liver cirrhosis (LC) and hepatocellular carcinoma (HCC) [1,2]. The term NAFLD is the counterpart of alcohol-related (-associated) liver disease (ALD) [3,4], which suggests that this clinico-pathological entity is heterogeneous, while excluding liver disease attributable to excessive alcohol consumption and other major etiologies such as viral hepatitis and autoimmunity. Most NAFLD is closely related to metabolic syndrome; therefore, NAFLD is now widely accepted as a liver manifestation of metabolic syndrome. Since liver is one of the key organs for the systemic regulation of metabolism [5], pathophysiological changes in the liver not only result in the progression of chronic liver disease, but also influence various systemic conditions, especially the development of atherosclerotic diseases [6].
The obvious and fundamental difference between NAFLD and ALD lies in the amount of alcohol consumption, and the clinical features of typical ALD are distinct from those of NAFLD, even though these two disease entities share similar pathological findings and pathophysiology. Recently, however, a considerable proportion of patients with ALD have also come to demonstrate comorbid metabolic syndrome as well [7]. Since the differential diagnosis of NAFLD and ALD is based solely on the level of alcohol intake, the combination of ALD and metabolic syndrome often causes confusion and difficulty in the clinical setting. Further, a portion of the patients likely to be diagnosed with NAFLD/NASH may be occasional drinkers, and/or even former heavy-drinkers. Given the lines of evidence indicating potential pathophysiological similarities between ALD and NAFLD/NASH, great attention should be paid to synergistic worsening of ALD in combination with metabolic syndrome.
NAFLD AND ALD IN ASIA
Metabolic syndrome is highly prevalent in industrialized Western countries [8-10], and is also a growing health problem in Asian countries [11,12]. Further, the susceptibility to obesity, diabetes, and cardiovascular disease (CVD) differs among various ethnic groups [13-15]. Thus, increasing attention is being paid to NAFLD/NASH, not only in the industrialized countries of the West but also in the East, with growing concern even in developing countries [16,17].
It is widely believed that the Western life-style, especially the Western diet, is one of the leading causes of metabolic syndrome and NAFLD [18]. In Asian countries, therefore, westernization of the diet and industrialization most likely have led to the increase in the prevalence of NAFLD [17]. The traditional Japanese diet (Washoku), which favors fish over meat dishes and rich in soybean products, is thought to be healthy and sustainable dietary patterns [19]. Thus, typical Japanese cuisine is believed to be protective against development of NAFLD.
On the other hand, Asian populations demonstrate higher susceptibility to diabetes, even though obesity levels are lower than in Western people [20,21]. It has also been reported that the prevalence of non-obese NAFLD is higher in Asian countries [22]. Thus, it seems likely that the prevalence of NAFLD/NASH will continue to rise if the westernization of life-style progresses further in developing countries in Asia.
In terms of alcohol intake, per capita consumption values are lower in Asian countries compared to developed countries in the West [23]. This disparity is partly due to ethnic differences in polymorphisms in ethanol (EtOH)-metabolizing enzymes [24]. In Asian populations, however, heterozygosity for the aldehyde dehydrogenase (ALDH) 2*2 allele, which result in lower ALDH2 activity, is highly prevalent at around 40–50% [25]. Such individuals need attention since they might suffer more severe liver injury following heavy drinking.
GENDER DIFFERENCES IN NAFLD AND ALD
There is overt gender difference in the development of ALD: women are more susceptible to ALD than men [26]. It is well-known that women develop more severe ALD even after consuming less alcohol over a shorter period [27]. The female predominance in ALD has also been demonstrated in animal models with equal EtOH loads on a body weight basis [28,29], indicating that the phenomenon is not a simple difference in body mass and/or liver volume as suggested in human studies [30,31]. In contrast, any gender difference in NAFLD is variable, and uncertain. Though some pioneering studies had suggested female predominance in NAFLD, the following population-based studies, especially based on the third NHANES data, have demonstrated a higher prevalence of NAFLD in male subjects [32]. In animal models, some reports indicated that male rodents develop more severe steatohepatitis than females [33,34].
With regard to the underlying mechanism of gender differences in ALD and NASH, it has been proposed that estrogen plays a pivotal role, possibly in different ways. It has been demonstrated that ovariectomy prevents liver injury following long-term EtOH administration in rodents, whereas estrogen replacement reverses the phenomenon, indicating that estrogen exacerbates alcohol-induced liver injury [29]. In contrast, estrogen has been suggested to play a preventive role in non-alcoholic models of steatohepatitis in rodents [33]. These findings are consistent with the clinical observations that women at young ages develop milder NAFLD than men, while NAFLD in women is more prevalent in those who past menopause [35,36].
POTENTIAL MECHANISTIC INSIGHTS IN THE PATHOGENESIS OF NAFLD AND ALD
Relationship between metabolic syndrome and fatty liver/steatohepatitis
In the relationship between metabolic diseases and NAFLD, causality may not be simply unidirectional. Epidemiological studies have suggested positive correlations between the prevalence of metabolic phenotypes and NAFLD/NASH, leading to the hypothesis that pathophysiological changes in the liver initiate systemic alterations in metabolism, thereby exaggerating atherosclerotic diseases. It is also conceivable that systemic micro-inflammation triggers insulin resistance, vascular damage, and NAFLD/NASH, simultaneously. To some extent, a fatty liver per se may be benign or even protective against systemic metabolic disorders. Fat accumulation in the liver is a physiological response to dietary overload, preventing over-flux of excess nutrients into the systemic circulation. Thus, it can be argued that fat accumulation in the liver, which is induced by hyperinsulinemia, would serve as a buffer to protect from, or delay, the onset of overt diabetes. The capacity of fat storage in the liver, therefore, may be critical for the progression of systemic metabolic disorders. Moreover, emerging lines of evidence suggest a possible sub-classification of NAFLD based on genetic variants, which would predict an atherosclerotic phenotype distinct from progressive liver disease. In summary, there is a close relationship between metabolic diseases and NAFLD/NASH, while the pathophysiological mechanisms remain to be explored in greater detail.
Genetic background in NAFLD and ALD
Since NAFLD is a heterogeneous amalgamation of fatty liver diseases, genetic predisposing factors seem to be diverse as is true for other metabolic syndrome phenotypes such as diabetes and dyslipidemia. The heritability of NAFLD has been estimated to be around 39% in a familial aggregation study [37], though the influence of environmental factors cannot be entirely excluded.
A genome-wide association study revealed that a single nucleotide polymorphism (SNP) of the patatin-like phospholipase domain-containing 3 (PNPLA3) gene is associated with susceptibility to NAFLD [38]. The I148M variant of the PNPLA3 gene is associated with the development of fatty liver in both alcoholic and nonalcoholic individuals. Although it is likely that the genetic variation of PNPLA3 is not associated with other manifestations of metabolic syndrome, there is a certain link between PNPLA3 gene polymorphisms and ethnicity [38,39]. Association of PNPLA3 variants with NAFLD has also been shown in Asian populations in China [40], Japan [41], and Korea [42,43]. Aside from PNPLA3, variants in other genes have been identified as genetic risk factors for NAFLD/NASH, including transmembrane 6 superfamily member 2 (TM6SF2) and apolipoprotein C (APOC3), etc [44].
Notably, PNPLA3 variants are also a significant genetic risk for ALD [45]. Though a variety of EtOH-metabolizing enzymes show gene polymorphisms, which are closely related to addictive phenotypes and the ability take in EtOH, PNPLA3 gene polymorphism has been shown to be the most critical risk for progressive organ injury.
Lipid dynamics, EtOH metabolism and formation of a fatty liver
In the process of fatty liver development, regardless of metabolic background or alcohol intake, impaired balance among uptake, synthesis, catabolism, and production/release results in fat accumulation in hepatocytes. A variety of regulatory molecules contribute to lipid dynamics in the liver (Fig. 1). Ethanol is mainly metabolized in hepatocytes through two major enzymes, alcohol dehydrogenase (ADH) and cytochrome P450 (CYP) 2E1. In addition to redox shift by EtOH metabolism, reactive oxygen species (ROS) generated by CYP2E1, as well as from damaged mitochondria, is involved in oxidative cellular injury [46]. Redox shift by increasing the NADH/NAD+ ratio is known to suppress gluconeogenesis. Inhibition of β-oxidation is also an important step in ethanol-induced lipid accumulation in hepatocytes (Fig. 2).
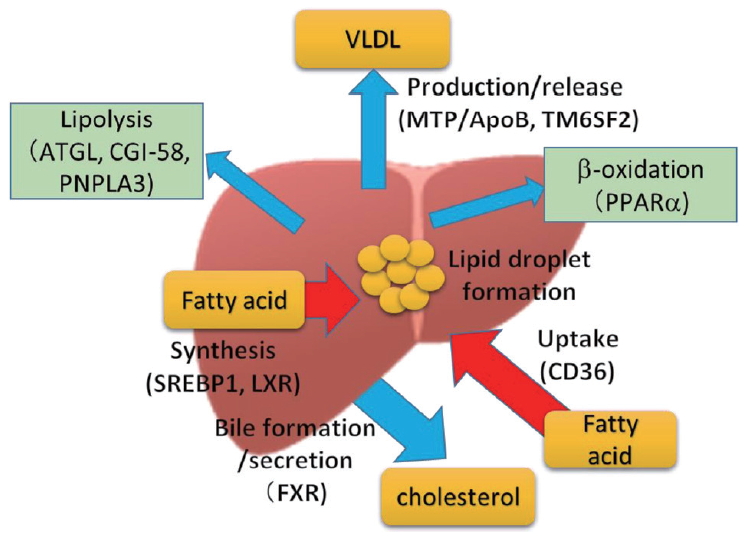
Impaired balance among uptake, synthesis, catabolism, and production/release results in fat accumulation in hepatocytes. VLDL, very low density lipoprotein; ATGL, adipose triglyceride lipase; CGI-58, comparative gene identification 58; PNPLA3, patatin-like phospholipase domain-containing 3; MTP, microsomal triglyceride transfer protein; ApoB, apolipoprotein B; TM6SF2, transmembrane 6 superfamily member 2; PPARα, peroxisome proliferator-activated receptor α; SREBP1, sterol regulatory element binding transcription factor 1; LXR, liver X receptor; CD36, cluster of differentiation 36; FXR, farnesoid X receptor.
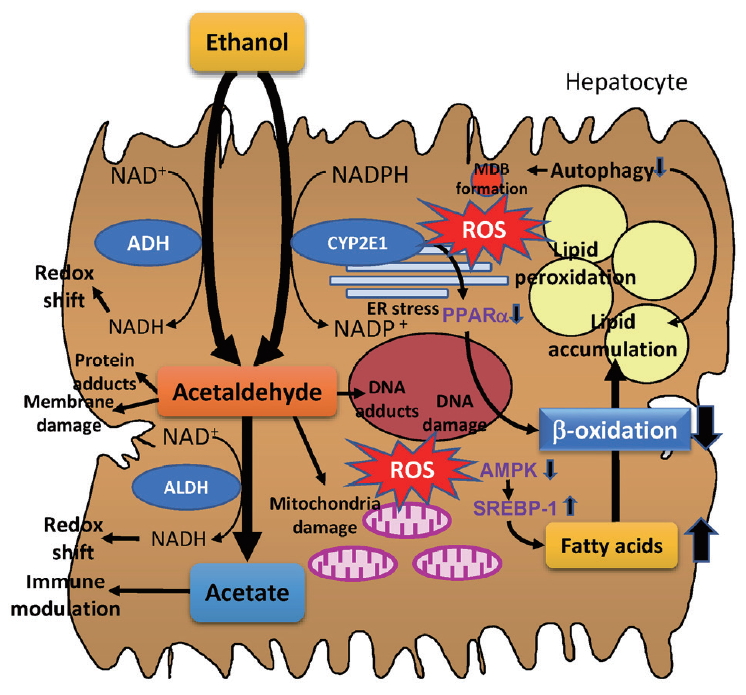
Ethanol metabolism and lipid accumulation in hepatocytes. NAD+, nicotinamide adenine dinucleotide+; NADPH, nicotinamide-adenine dinucleotide phosphate; MDB, mallory denk body; ADH, alcohol dehydrogenase; CYP2E1, cytochrome P450 2E1; ROS, reactive oxygen species; ER, endoplasmic reticulum; ALDH, aldehyde dehydrogenase; AMPK, AMP-activated protein kinase; SREBP-1, sterol regulatory element binding transcription factor-1.
Cellular stress responses in NAFLD and ALD
Cellular stress responses protect the cell, the organ and/or organism from toxic stimuli; however, inappropriate and/or overzealous responses contribute to disease pathogenesis. Indeed, there is significant crosstalk between stress responses and the innate immune response that may contribute to the initiation and progression of disease pathogenesis. The biochemical impact of steatohepatitis drives alterations in substrate supply and metabolism in the liver. For instance, lysosomal-degradation of unnecessary or dysfunctional cellular components is key not only for maintaining cellular energy levels during starvation, but also for an adequate response of this organ to stressors. ALD/NASH both impair autophagy [47], which is implicated in the pathogenesis of these diseases.
The endoplasmic reticulum (ER) is a multifunctional organelle required for the regulation of calcium homeostasis, lipid metabolism, and protein synthesis. A number of cellular stress conditions lead to the accumulation of unfolded or misfolded proteins in the ER and disruption of ER homeostasis, which can trigger ER stress. ER stress activates the unfolded protein response (UPR). The UPR pathway includes induction of several molecular chaperones that restore cellular homeostasis by promoting the folding or degradation of unfolded proteins; however, if ER stress is prolonged or too severe, the signaling switches from pro-survival to pro-death, leading to ER stress-induced apoptosis. Several studies have shown that ER stress contributes to the development of ALD [48].
To investigate the comorbidity of ALD and metabolic syndrome, we have recently applied the mouse model of chronic-binge EtOH liver injury (NIAAA model) to obese KK-Ay mice [49]. Chronic-plusbinge EtOH intake induced massive hepatic steatosis along with hepatocyte apoptosis and inflammation, and increased ER stress markers including binding immunoglobulin protein (BiP), unspliced and spliced forms of X-box-binding protein-1 (uXBP1 and sXBP1, respectively), inositol trisphosphate receptor (IP3R), and C/EBP homologous protein (CHOP), and also enhanced the oxidative stress markers heme oxygenase-1 and 4-hydroxynonenal. Administration of the chemical chaperone 4-phenylbutyric acid during chronic EtOH exposure ameliorated steatohepatitis after chronic-binge EtOH, and completely inhibited both ER and oxidative stress markers. These findings indicated that binge EtOH intake after chronic consumption induces massive ER stress-related oxidative stress followed by liver injury, and inhibition of ER stress using a chemical chaperone is a potential preventive therapy for alcoholic liver injury especially in obese subjects.
Gut microbiota in steatohepatitis due to metabolic syndrome and alcohol
Increasing attention has been paid to the gut microbiota in human health and disease [50]. Indeed, the gut microbiota is dynamically altered by dietary factors, lifestyle, and alcohol intake. Gut microbiota-dependent activation of hepatic innate immunity is important in the pathogenesis of steatohepatitis caused by both alcohol and metabolic syndrome (Fig. 3). Chronic alcohol exposure, as well as dietary overload, compromises gut barrier function causing increases in intestinal permeability, thereby aggravating translocation of bacterial products into the portal blood [51]. Pathogen-associated molecular patterns (PAMPs) derived from gut microbiota elicit production and release of inflammatory cytokines through multiple innate immune-signaling pathways, resulting in the exacerbation of steatohepatitis [52].
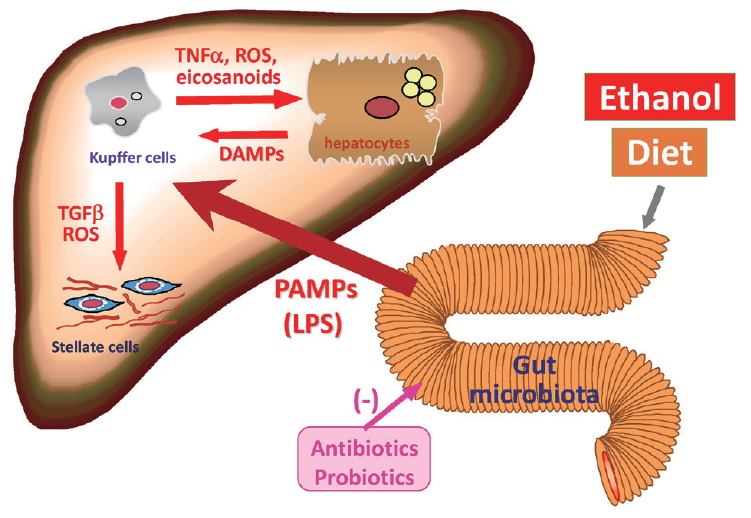
Gut microbiota-dependent activation of hepatic innate immunity caused by both alcohol and metabolic syndrome in the pathogenesis of steatohepatitis. TNF, tumor necrosis factor; ROS, reactive oxygen species; DAMP, damage- associated molecular pattern; PAMP, pathogen-associated molecular pattern; LPS, lipopolysaccharide.
We investigated the alteration in the small intestinal microbiota profile following chronic EtOH feeding in KK-Ay mice, and the effect of rifaximin (RFX) on liver injury following chronic/binge administration of EtOH [53]. Treatment with RFX significantly suppressed hepatic steatosis, as well as the increase in oxidative stress and inflammatory cytokines, in KK-Ay mice given EtOH-feeding/binge. Chronic EtOH feeding increased the net amount of small intestinal bacteria, but RFX did not prevent this increase. At the bacterial order level, however, EtOH dramatically increased the relative abundance of the Erysipelotrichales, whose increase in RFX-treated mice was drastically substituted for by the Bacteroidales. Small intestinal bacterial overgrowth (SIBO) and leaky gut result in the increase in bacteria-derived substances such as endotoxin (lipopolysaccharide [LPS]) in the portal blood, which elicit innate immune responses in the liver. Thus, it has been postulated that the modulation of small intestinal microbiota is critical for the prevention of alcoholic liver injury in the context of comorbid metabolic syndrome.
More recently, it has been demonstrated that Candidalysin, a fungal exotoxin, also contributes to the exacerbation of alcoholic hepatitis [54]. It has been reported that fecal levels of Candida albicans and endothelin-converting enzyme 1 are increased in patients with alcoholic hepatitis [55]. Candidalysin enhances ethanolinduced liver disease and is associated with higher mortality in mice. Indeed, Candidalysin damages hepatocytes in a dose-dependent manner. Moreover, Candidalysin is also associated with mortality and the severity of liver disease in patients with alcoholic hepatitis. The variety of bacterial and fungal components and products closely associated with the pathophysiology of steatohepatitis merit deep interest.
Innate immunity and ALD/NAFLD
It has been shown that metabolic reprogramming of hepatic cells during steatohepatitis is not relegated solely to hepatocytes, but hepatic macrophages also undergo metabolic reprogramming during ALD/NAFLD, which may contribute to polarizing the phenotype of the macrophages to a proinflammatory state. Importantly, PAMPs released from the GI tract activate the innate immune response in liver (Fig. 3). Indeed, innate immune responses appear to be primed for stimulus by PAMPs during ALD/NAFLD. Liver also contains other types of innate immune cells, including natural killer (NK) and NKT cells [56]. Both EtOH and diet alter the expression patterns of hepatic NK and NKT cells, which dysregulates not only the Th1/Th2 balance in hepatic microenvironment, but also immune surveillance against carcinogenesis [57]. Therefore, maintaining intestinal barrier integrity and the balance of gut microbiota appears to be key in preventing ALD/NAFLD.
ALD/NAFLD AND HCC
Epidemiological surveys have revealed that the proportion of HCC based on non-viral chronic liver diseases is gradually increasing in Japan, although the majority of HCC develops in patients with chronic viral hepatitis C and B [58]. However the contribution of ALD is far from negligible, accounting for almost half of non-viral HCC. Since metabolic syndrome-related NASH appears to be increasing, NASH can be predicted to take the place of viral hepatitis as the major cause of HCC even in Asian countries including Japan in the foreseeable future.
Nowadays, we sometimes encounter patients with NASH bearing large HCC tumors, due to the lack of proper screening strategies [59]. In screening fatty liver individuals for HCC, one of the critical disadvantages is the poor diagnostic value of ultrasound examination. Magnetic resonance imaging is a preferable technique for detection of tumorous lesions in fatty liver; however, it is less cost-effective and not suitable for mass-screening.
From the viewpoint of pathophysiology, the mechanisms underlying hepatic carcinogenesis in steatohepatitis most likely involve metabolic abnormalities including altered sensitivity to insulin, excessive cellular stress/damage responses, and impaired immune surveillance. Further, emerging roles for bacterial metabolites including short-chain fatty acids and secondary bile acids in metabolic modulation and carcinogenesis have been proposed [60,61], suggesting potential prophylactic/therapeutic targets.
EXTRAHEPATIC CARCINOGENESIS IN ALD AND NAFLD/NASH
Besides HCC, both alcohol and metabolic syndrome have also been demonstrated increase the risk of cancer development in extrahepatic organs. The affected organs, however, show certain differences and similarities between ALD and metabolic syndrome-based NAFLD. ALD patients, especially those drinking beverages of higher alcohol content, often develop esophageal cancer, as well as head and neck tumors [62,63]. In contrast, esophageal cancer is less frequent in NAFLD individuals, while increases in waist circumference is associated with increased risk of Barrett cancer [64]. On the other hand, both alcohol and diabetes are overt risk factors for pancreatic cancer [65]. Similarly, both higher alcohol intake and obesity/NAFLD have been reported as risk factors for cancers of the colon and breast [66,67]. It is therefore important to pay attention to systemic screening for cancer in patients of both ALD and NAFLD/NASH.
CONCLUSIONS
NAFLD is now the most prevalent chronic liver disease, especially in developed countries worldwide. NASH, a progressive form of NAFLD, also gives rise to HCC in its advanced stages. It is of note that alcohol intake, even in moderate levels, synergistically worsens metabolic steatohepatitis. Since ALD and NASH share a common pathophysiological basis involving the gut-liver axis, alterations in the gut microbiota (dysbiosis) and subsequent changes in a variety of microbiota-derived components/metabolites most likely contribute to the synergistic actions of alcohol and metabolic factors in the progression of steatohepatitis and carcinogenesis.
Acknowledgements
The authors thank Dr. Robert F. Whittier (Project Professor, Division of Medical Education, Juntendo University School of Medicine) for English proofreading and critical advice on the manuscript.
Notes
Conflicts of Interest
The authors have no conflicts to disclose.
Abbreviations
ADH
alcohol dehydrogenase
ALD
alcohol-related liver disease
ALDH
aldehyde dehydrogenase
APOC3
apolipoprotein C
BiP
binding immunoglobulin protein
CHOP
C/EBP homologous protein
CVD
cardiovascular disease
CYP
cytochrome P450
ER
endoplasmic reticulum
EtOH
ethanol
HCC
hepatocellular carcinoma
IP3R
inositol trisphosphate receptor
LC
liver cirrhosis
LPS
lipopolysaccharide
NAFL
nonalcoholic fatty liver
NAFLD
nonalcoholic fatty liver disease
NASH
nonalcoholic steatohepatitis
NK
natural killer
PAMP
pathogen-associated molecular pattern
PNPLA3
patatin-like phospholipase domain-containing 3
RFX
rifaximin
ROS
reactive oxygen species
SIBO
small intestinal bacterial overgrowth
SNP
single nucleotide polymorphism
sXBP1
spliced forms of X-box-binding protein-1
TM6SF2
transmembrane 6 superfamily member 2
UPR
unfolded protein response
uXBP1
un-spliced of X-box-binding protein-1