Roles of the complement system in alcohol-induced liver disease
Article information

Abstract
Alcohol-induced liver disease (ALD) is a complex disorder, with a disease spectrum ranging from steatosis to steatohepatitis, cirrhosis, and hepatocellular carcinoma. Although the pathogenesis of ALD is incompletely understood and currently no effective drugs are available for ALD, several lines of evidence suggest that complement activation and oxidative stress play crucial roles in the pathogenesis of ALD. Complement activation can regulate the production of ROS and influence oxidative stress in ALD. Precise regulation of the complement system in ALD may be a rational and novel avenue to postpone and even reverse the progression of disease and simultaneously promote the repair of liver injury. In this mini-review, we briefly summarize the recent research progress, especially focusing on the role of complement and oxidative stress-induced transfer RNA-derived fragments, which might help us to better understand the pathogenesis of ALD and provide aid in the development of novel therapeutic strategies for ALD.
OVERVIEW OF THE COMPLEMENT SYSTEM
Complement activation is a key part of the innate immune system, which plays a crucial role in the defense against microbial infections, healing of the host, and disposal of the products of inflammatory injury [1-3]. The complement system consists of complement components, receptors, and regulatory factors. Most of the serum complement components are produced in the liver, and many complement receptors (CRs) are expressed in liver Kupffer cells and hepatic stellate cells [4-6]. According to the recognition of different molecules, complement activation can be divided into three main pathways: the classical pathway (CP), the alternative pathway (AP), and the lectin pathway (LP) (Fig. 1) [7]. The CP is triggered by C1q recognition of antibodies bound to antigens or microbial surfaces, resulting in the activation of C1r and C1s. The activated C1s then cleaves C4 and C2 to form C3 convertase (C4bC2b). The LP is similar to the CP; they share C3 convertase (C4bC2b). However, the LP does not rely on antibodies to identify pathogenic components but uses mannan binding lectin (MBL) and ficolins to identify sugars or N-acetylated groups on the surface of pathogens. In response, MBL-associated serine proteases (MASP1/MASP3 and MASP2) are activated [8]. The activated MASP1/MASP3 and MASP2 can cleave C2 and C4 to form C4bC2b [9,10]. The AP is different from the CP and LP; the AP is initiated by C3 itself. In the first step, the spontaneous hydrolysis of the thioester bond within C3 takes place, resulting in the formation of C3(H2O). Then, C3(H2O) recruits factor B (FB) to form a complex, which can be further cleaved by factor D (FD), leading to the formation of C3(H2O)Bb. C3(H2O)Bb is also a C3 convertase enzyme complex, which cleaves C3 to C3a and C3b. C3b can be added to FB and cleaved by FD, resulting in the formation of the C3 convertase C3bBb, which acts as an amplification loop of the complement system [11,12].
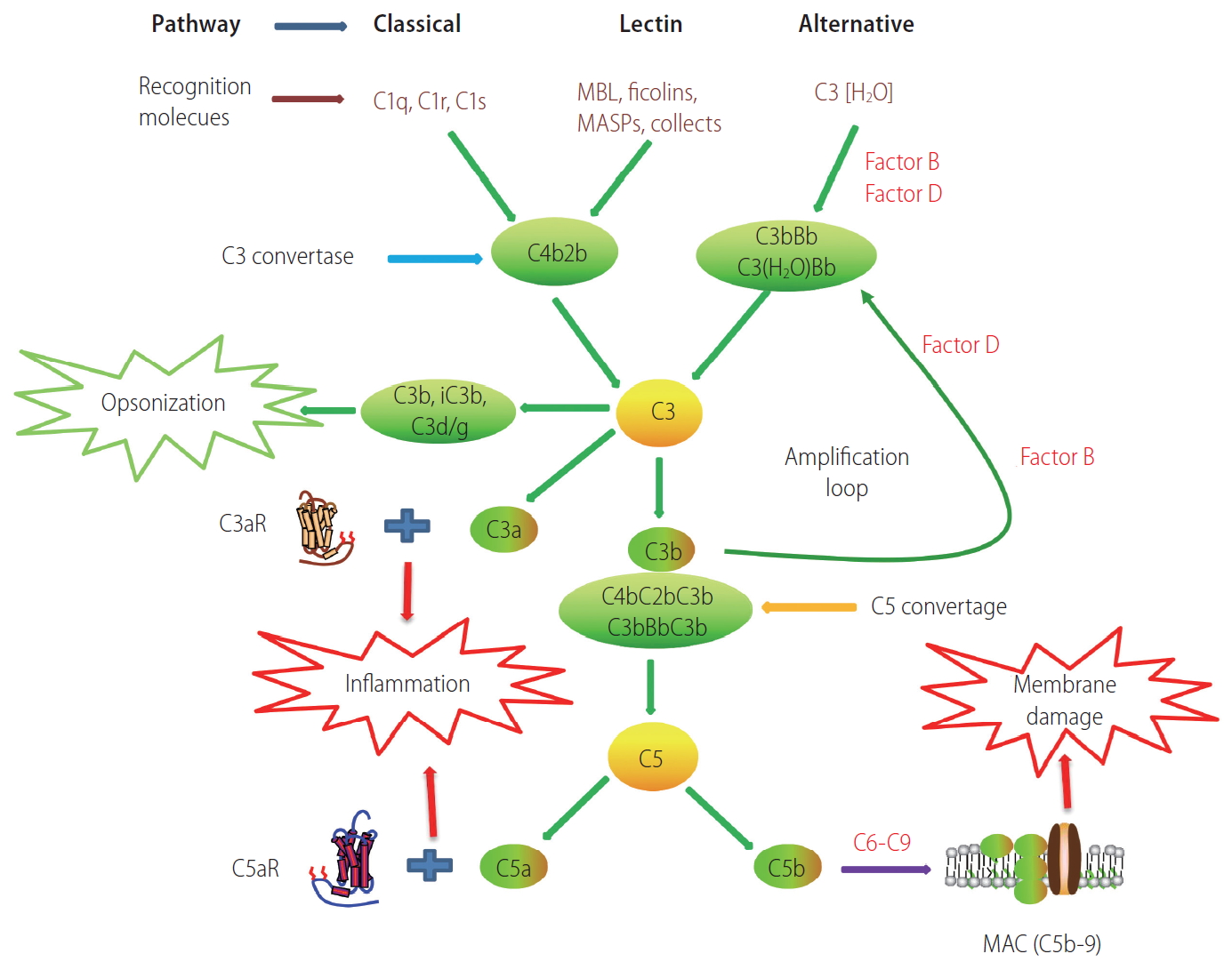
Schematic overview of the complement cascade illustrating three activation pathways (classical, lectin, and alternative) and summarizing the biological effects of complement activation products. MAC, membrane attack complex.
The above three pathways produce C3 convertase and C5 convertase, which can cleave the central complement components C3 and C5, respectively, and generate effector molecules [13]. Anaphylatoxins, including C3a and C5a, are important potent pro-inflammatory molecules that attract and activate immune cells. Opsonin C3b (or C4b) is deposited on the surface of pathogens or injured cells, and mediates immune adhesion and opsonization or induces cytolysis through the membrane attack complex (MAC). The MAC consists of C5b, C6, C7, C8, and C9, which can directly lyse targeted pathogens or damaged autologous cells [14,15].
In recent years, the comprehensive evaluation of complement function has shown that the complement system not only plays a traditional role in the immune process but also participates in a variety of important physiological functions, such as lipid metabolism, the inflammatory response, coagulation, angiogenesis, and tissue repair [2,10,16-20]. A growing number of studies have revealed that the complement system also plays essential roles in the progress of alcoholic liver disease (ALD) [21-23]. In this mini-review, we used five search engines, that is, MEDLINE, PubMed, EMBASE, Web of Science, and Biosos Preview, and we screened out 218 papers in this field. We will discuss the recent findings on the role of complement activation and regulation and oxidative stress in the progression of ALD, which might help us to better understand the pathogenesis of ALD and provide some novel potential therapeutic strategy for ALD.
COMPLEMENT AND ALCOHOL-INDUCED LIVER DISEASE
Studies have reported that the complement system is activated in ALD and participates in different stages of its pathogenesis, including alcoholic fatty liver disease (AFLD), alcoholic hepatitis (AH), alcoholic hepatic fibrosis, and cirrhosis (Table 1) [21-23]. In this mini-review, the role and mechanism of the complement system in different stages of ALD are briefly summarized, especially focusing on the recent relevant progress.
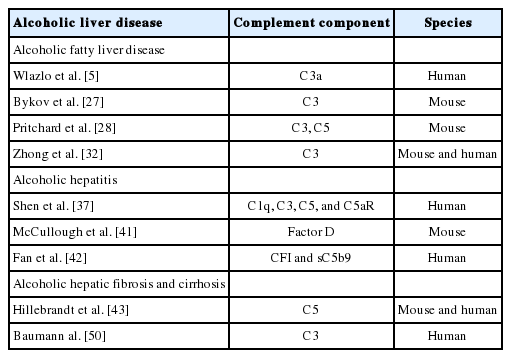
Important research of the complement system in alcoholic liver disease
The complement system participates in AFLD
The liver is a predominant organ for the metabolism of alcohol and lipids [24]. It is generally accepted that in the pathogenesis of AFLD, alcohol, as the “first strike,” causes the disorder of lipid metabolism and the heterotopic deposition of triglycerides in hepatocytes. The reaction between reactive oxygen species (ROS) and accumulated triglycerides in hepatocytes is lipotoxic and acts as the “second strike” in the AFLD process [25]. However, how the complement system is involved in this process remains unclear.
Barry and McGivan [22] reported that acetaldehyde, an intermediate product of alcohol metabolism, can bind to hepatocyte plasma membranes, resulting in the activation of C3 by causing a structural change in the surface of the plasma membrane. A previous study in rats revealed that the expression of complement components (C1, C3, and C8) was elevated, while the expression of complement receptor 1 (CR1)-related protein y (Crry) and CD59 was reduced after long-term alcohol feeding [26]. Wlazlo et al. [5] showed that the serum level of C3a, an indicator of C3 activation, was associated with liver steatosis and hepatocellular injury in individuals who are chronic alcohol abusers. In mouse models of ethanol feeding, Bykov et al. [27] found that C3+/+ mice developed AFLD, while C3–/– mice had reduced lipid accumulation in the liver. In a study of the role of C3 in AFLD, the authors suggested that C3a binding to the receptor C3aR, which is highly expressed on Kupffer cells, can promote the inflammatory response and stimulate the release of the inflammatory cytokine tumor necrosis factor-α (TNF-α). TNF-α participates in AFLD, either directly or indirectly, through the induction of insulin resistance [5]. Pritchard et al. [28] also proved that C3–/– mice did not develop AFLD or increase triglycerides in the circulation, but the levels of serum alanine aminotransferase (ALT) and inflammatory cytokines were significantly higher. On the contrary, C5–/– mice had increased hepatic triglycerides and developed AFLD, whereas they did not show increased levels of ALT or inflammatory cytokines after ethanol feeding. Another study showed that more cholesterol was deposited in the liver of C5–/– mice than in the liver of C5+/+ mice, and C5–/– mice had higher levels of serum triglycerides and cholesterol than C5+/+ mice [23]. These results show that C3 and C5 play different roles in the development of AFLD. However, the mechanisms by which the complement system is involved in AFLD are still incompletely understood.
Ethanol consumption increases the generation of ROS and decreases the antioxidant activity, leading to the increase of oxidative stress. Increased oxidative stress from ethanol exposure is a critical element for the pathogenesis of ALD [29]. Growing evidence suggests that the interaction between complement activation and oxidative stress plays crucial roles in ALD [29-31]. Under basal conditions, the anaphylatoxin C3a and C5a receptors are highly expressed in Kupffer cells. Ethanol-induced complement activation results in the generation of the anaphylatoxins C3a and C5a, which bind to the receptors on Kupffer cells, and subsequently increases the expression of inflammatory cytokines and releases the ROS, contributing to ALD [28]. However, the mechanisms underlying the interaction between complement activation and oxidative stress in ALD remain unclear. A recent study found that C3a and its degraded form, C3a-des-Arg (also known as acylation stimulating protein [Asp]), contribute to the pathogenesis of AFLD. Asp binding to its receptor C5aR2 promotes the expression of cytochrome P450 family 2, subfamily E, polypeptide 1, which induces the production of ROS. The induced oxidative stress subsequently leads to the increased expression of glycine tRNA-derived fragments (Gly-tRFs). Gly-tRF antisense inhibitor treatment can prevent the development of AFLD by reducing fatty acid synthesis and increasing fatty acid oxidation through regulating the SIRT1 signaling pathway in ethanol-fed mice. This study bridges the knowledge gap between the complement system, oxidative stress, and steatosis through elucidating the role of Gly-tRFs in ALD [32].
The complement system participates in alcoholic hepatitis
Different from AFLD, except for the deposition of triglycerides in hepatocytes, AH is also characterized by the infiltration of inflammatory cells, elevated inflammatory cytokine levels, increased serum transaminase levels, and liver injury. Previous studies have shown that inflammatory cytokines play an important role in the occurrence and development of ALD [33,34]. In AH, inflammatory cytokine levels were elevated, leading to liver cell dysfunction and continued tissue damage [35]. The complement system is also associated with the pathogenesis of AH. Cohen et al. [36] reported that C1q deficiency abolishes the activation of the CP, reduces the expression of inflammatory cytokines, and attenuates liver tissue injury. These results were further confirmed in a study of an alcoholic liver model in mice by Smathers et al. [34]. Moreover, in AH patients, C1q and C5 levels were significantly increased, and the expression of C5aR was also upregulated in the Mallory-Denk body forming cells, which are the major features of ongoing inflammation in AH [37,38]. These studies showed that complement activation is involved in the occurrence of AH and the impairment of liver function. The underlying mechanism may be that there is a crosstalk between toll-like receptors (TLRs) and C3aR and C5aR in AH [21,37,39,40]. TLRs belong to the pattern recognition receptor family, which can bind to damage-associated molecular patterns or pathogen-associated molecular patterns (PAMPs). PAMPs mainly refer to bacterial metabolites, including bacterial lipopolysaccharide, peptidoglycan, RNA, and DNA, which can reach the liver upon alcohol-induced intestinal microecology imbalance, subsequently initiating the innate immune response and inducing the expression of inflammatory cytokines (such as TNF-α and interleukin-6). In addition, PAMPs increase lipid deposition and inflammatory infiltration in hepatocytes. McCullough et al. [41] found that complement FD promotes the clearance of apoptotic cells and maintains tissue homeostasis in AH and suggested that inappropriate complement activation likely delays the clearance of apoptotic cells and impairs healing/recovery in ALD. Fan et al. [42] found that complement activation is expected to be a diagnostic and prognostic indicator for patients with AH. However, the detailed mechanisms of complement function in AH need to be further clarified.
The complement system participates in alcoholic hepatic fibrosis and cirrhosis
Hepatic fibrosis is an early event of cirrhosis. Alcoholic hepatic fibrosis is a process of damage and repair of the liver after long-term alcohol exposure, accompanied by the excessive accumulation and rearrangement of the extracellular matrix, the development of AH, and the formation of pseudolobules. These pathological changes cause damage to the liver structure and ultimately progress to liver cirrhosis. A previous study showed that the expression of the C5 gene is involved in liver fibrogenesis, and inhibition of the C5aR1 attenuates liver fibrosis in mice [43]. Moreover, C5 has a causal role in fibrogenesis across species between humans and mice. A number of studies have confirmed a decrease in the level of serum complement proteins, including C1, C3, C4, C5, and FB, in patients with alcoholic cirrhosis [44-47]. Other studies further revealed that serum concentrations of C3, C4a, and C5a had a negative relationship with liver fibrosis stages and the Child-Pugh score [48-50]. These studies show that the levels of complement components may be an indicator of liver fibrosis or cirrhosis stage [51]. There are two possible mechanisms: the reduction of complement synthesis after severe liver injury and excessive complement depletion [52]. However, the results of two studies showed some discrepancies; the serum complement concentration was normal, and no significant differences were observed in patients with alcoholic cirrhosis [53,54]. Further studies are required to explore the mechanisms underlying the role of the complement system in the occurrence and development of advanced liver disease, as complement regulation may be a potential strategy for reversing the progress of liver fibrosis.
COMPLEMENT REGULATION IN ALCOHOLIC LIVER DISEASE
In physiological conditions, there is a delicate balance between complement activation and regulation. Insufficient regulation or excessive activation can result in a large variety of disorders [55]. The host expresses a variety of both fluid-phase and membranebound inhibitory proteins to regulate the location and activity of complement factors. Soluble inhibitors of the complement system include C1 inhibitor, C4 binding protein, factor I, factor H (FH), and MBL-associated protein of 44 kilodalton, which can be distributed throughout the body with the blood [56]. These soluble inhibitors have the characteristics of systematic regulation, which easily lead to immune regulation imbalance and lack of targeting. Membrane-bound inhibitory proteins are anchored on the membrane surface of specific cells to protect the host from complement attack. These membrane-bound inhibitors can be divided into two categories. The first includes inhibitors of the formation of C3 convertases, including CR1, decay accelerating factor, and membrane cofactor protein. Crry is expressed in rodents, which is similar to human CR1 in structure and function. The other is represented by CD59, which competes to combine with C8 and C9, further inhibiting MAC formation [57].
Tissue targeting is a promising complement regulation strategy, which provides a safer and considerably more effective therapeutic approach compared with a systemic (untargeted) complement inhibition. One such targeting approach involves linking a complement inhibitor to a fragment of CR2 that recognizes C3 activation products deposited at sites of complement activation (Fig. 2). Complement inhibitors such as Crry, FH, or CD59 will go directly to the complement-attacked tissue after systemic infusion and locally inhibit complement without compromising the systemic complement function [55]. CR2-Crry, a site-targeted complement inhibitor, can suppress C3 activation through inhibition of C3 convertase, resulting in a reduction in the levels of C3a, C3b, and Asp. A recent study showed that suppression of C3 activation can protect against the injury of steatosis and inflammation in ALD [32]. Therefore, targeted complement inhibitors may become a potential drug to protect against both infiammatory response syndrome and hepatic steatosis for AFLD patients (Fig. 3) [32].

Schematic of CR2 site-targeted complement inhibitors. The CR2 moiety of the fusion protein binds the C3 degradation products iC3b, C3dg, and C3d that are covalently attached at sites of complement activation. Complement inhibitory constructs that have previously been prepared and characterized are CR2-Crry (murine inhibitor of C3 activation), CR2-CR1 (human inhibitor of C3 activation), CR2-fH (murine and human inhibitor of the alternative complement pathway), and CR2-CD59 (murine and human inhibitor of MAC formation). F1, factor 1; SP, serum protease; CR1/2, complement receptor 1/2; Crry, complement receptor-1 related protein y.
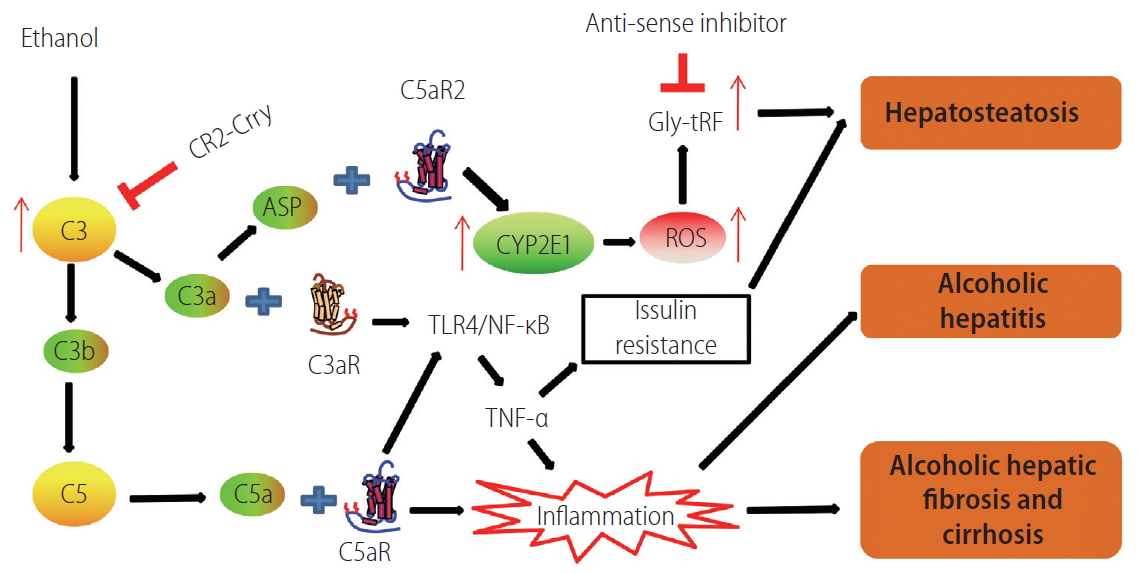
Schematic graph showing that the complement system participates in the development of alcohol-induced liver disease. C3 convertase cleaves C3 to form C3a and C3b. ASP is the degraded product of C3a. C3b is an important component of C5 convertase. CR2-Crry is a site-targeted complement inhibitor that blocks all complement pathways at the C3 activation step. When anaphylatoxins C3a and C5a bind with their respective receptors, the TLR4/NF-κB pathway can be activated, resulting in the release of the inflammatory cytokine TNF-α, which participates in AFLD, either directly or indirectly, through the induction of insulin resistance. C3a and ASP bind to their receptor C5aR2 to promote the expression of Cyp2E1, which induces the production of ROS. The induced oxidative stress subsequently leads to the increased expression of Gly-tRF, which leads to AFLD via regulating the SIRT1 signaling pathway. The inflammatory response contributes to the development and progression of ALD. CR2, complement receptor 2; Crry, complement receptor 1-related protein y; ASP, acylation stimulating protein; Cyp2E1, cytochrome P450 family 2, subfamily E, polypeptide 1; GlytRFs, glycine tRNA-derived fragments; ROS, reactive oxygen species; TLR, toll-like receptor; TNF-α, tumor necrosis factor-alpha; AFLD, alcoholic fatty liver disease; ALD, alcohol-induced liver disease.
According to the specific structure of complement molecules, complement mutants and complement peptides can also be used to regulate complement activity [58,59]. Moreover, the crosstalk between the complement, coagulation, and fibrinolysis systems also plays an important role in complement regulation. As an example, thrombomodulin participates in complement regulation through two mechanisms. Thrombomodulin can enhance factor I-mediated inactivation of C3b on the cell surface. Moreover, thrombomodulin also accelerates the inactivation of anaphylatoxins C3a and C5a by regulating the activation of procarboxypeptidase B [60,61]. These strategies may be also applicable to the treatment of ALD.
FUTURE PERSPECTIVE
Recent advances in complement research have shown that the complement system acts an important regulator for each stage of ALD. The precise regulatory mechanisms of the complement system in the occurrence and development of ALD need to be further explored. A better understanding of the mechanisms by which the complement system affects ALD may help us identify novel therapeutic targets and provide a potential therapeutic approach to inhibit or reverse the progression of ALD. Based on the advances of the present research, targeted complement inhibitors and antisense inhibition of Gly-tRF may provide a therapeutic strategy for treating human ALD.
Acknowledgements
This work was supported in part by National Natural Science Foundation of China (81771674 and 81660103); The 111 Project (D17011); Guangxi Key Research and Development Plan (2018AD03001); Guangxi BaGui Scholars; Molecular Medicine of Liver Injury and Repair Collaborative Innovation Center; Guangxi Key Laboratory of Molecular Medicine in Liver Injury and Repair (GXLIRMMKL-201806); and Special projects of local science and technology development guided by the central government (Declaration No 001176111039).
Notes
Authors’ contributions
He SQ designed research; Zhou Y, Zhong FD and Yuan GD searched the relevant literatures; and Zhou Y wrote the review; Zhong FD, Yuan GD and He SQ revised the review.
Conflicts of Interest
The authors have no conflicts to disclose.
Abbreviations
AFLD
alcoholic fatty liver disease
AH
alcoholic hepatitis
ALD
alcoholic liver disease
ALT
alamine aminotransferase
AP
alternative pathway
Asp
acylation stimulating protein
CP
classical pathway
CR1
complement receptor 1
Crry
complement receptor 1-related protein y
CRs
complement receptors
FB
factor B
FD
factor D
FH
factor H
Gly-tRFs
glycine tRNA-derived fragments
LP
lectin pathway
MAC
membrane attack complex
MBL
mannan binding lectin
PAMPs
pathogen-associated molecular patterns
ROS
reactive oxygen species
TLRs
toll-like receptors
TNF-α
tumor necrosis factor-α