Leaky gut-derived tumor necrosis factor-α causes sarcopenia in patients with liver cirrhosis
Article information

Sarcopenia is frequently seen in patients with liver cirrhosis and an independent risk factor for poor prognosis [1-5]. Sarcopenia is an important therapeutic target. However, its pathogenesis remains unclear, and a therapeutic strategy has not been established in patients with liver cirrhosis [1,5]. A recent study by Han et al. [6] investigated the association of liver cirrhosis-related systemic inflammation with sarcopenia in a rat model of liver cirrhosis. They found that tumor necrosis factor-α (TNF-α) was associated with the expression of intestinal tight junction proteins, muscular myostatin, and sarcopenia in a rat model of liver cirrhosis. Furthermore, they reported that treatment with rifaximin caused muscle hypertrophy with a reduction in both serum TNF-α levels and expression of muscular myostatin in a rat model of liver cirrhosis. Thus, they revealed that 1) TNF-α is involved in the pathogenesis of sarcopenia, and 2) rifaximin is a possible therapeutic strategy for liver cirrhosis-related sarcopenia through the downregulation of TNF-α.
Aging and physical inactivity are the main mechanisms underlying the development of sarcopenia [1]. Besides these factors, various liver-related metabolic dysfunctions are involved in the pathogenesis of sarcopenia in patients with liver cirrhosis [1,5,7]. The metabolic dysfunctions are depletion of branched-chain amino acids, carnitine, vitamin D, testosterone, and hyperammonemia [1,5,7]. In patients with liver cirrhosis, chronic inflammation is associated with development of various complications including ascites. However, information is limited on the association between inflammatory cytokines and sarcopenia. Han et al. [6] found a significant negative correlation of serum TNF-α level with muscle weight and myofiber diameter in a rat model of liver cirrhosis. Furthermore, they found that serum TNF-α levels were significantly higher in patients with sarcopenia than in those without sarcopenia [6]. Shiraki et al. [8] previously reported that, in patients with liver cirrhosis, elevated serum TNF-α levels were associated with malnutrition. In addition, TNF-α has been reported to promote myosin heavy-chain degradation and apoptosis of muscle fibers, leading to muscle atrophy [9,10]. These findings suggest that upregulation of TNF-α is important in the pathogenesis of sarcopenia in patients with liver cirrhosis.
In the liver, TNF-α is mainly released from Kupffer cells and hepatic stellate cells by stimulation of intestinal bacteria and their products, including lipopolysaccharide [11]. Therefore, increased intestinal permeability seems to be an upstream event for the upregulation of serum TNF-α levels in patients with liver cirrhosis. In fact, a variety of basic and clinical studies have implicated that gut dysbiosis affects the intestinal epithelial barrier and leads to translocation of gut contents to the liver and beyond [12,13]. Intestinal permeability is regulated by intercellular adhesion complexes called tight junctions [14]. Han et al. [6] demonstrated that expression of tight junction proteins such as occludin and zonula occludens-1 (ZO-1) in the intestine were inversely correlated with serum TNF-α levels in a rat model of liver cirrhosis. These findings are in line with results of previous studies. Intestinal expression of claudin-1 and occludin, tight junction proteins, has been reported to be associated with endotoxin levels in a rat model of liver cirrhosis [15]. Intestinal expression of claudin-1 has also been reported to be reduced and inversely correlated with endotoxin concentrations in patients with liver cirrhosis [16]. Furthermore, Han et al. [6] first demonstrated that intestinal expression levels of occludin and ZO-1 were positively correlated with muscle weight and myofiber diameter. Taken together, disruption of the intestinal tight junction may be responsible for the influx of lipopolysaccharide into the liver. Lipopolysaccharide stimulates Kupffer cells and hepatic stellate cells, leading to releasing TNF-α. Upregulated TNF-α causes sarcopenia in patients with liver cirrhosis (Fig. 1).
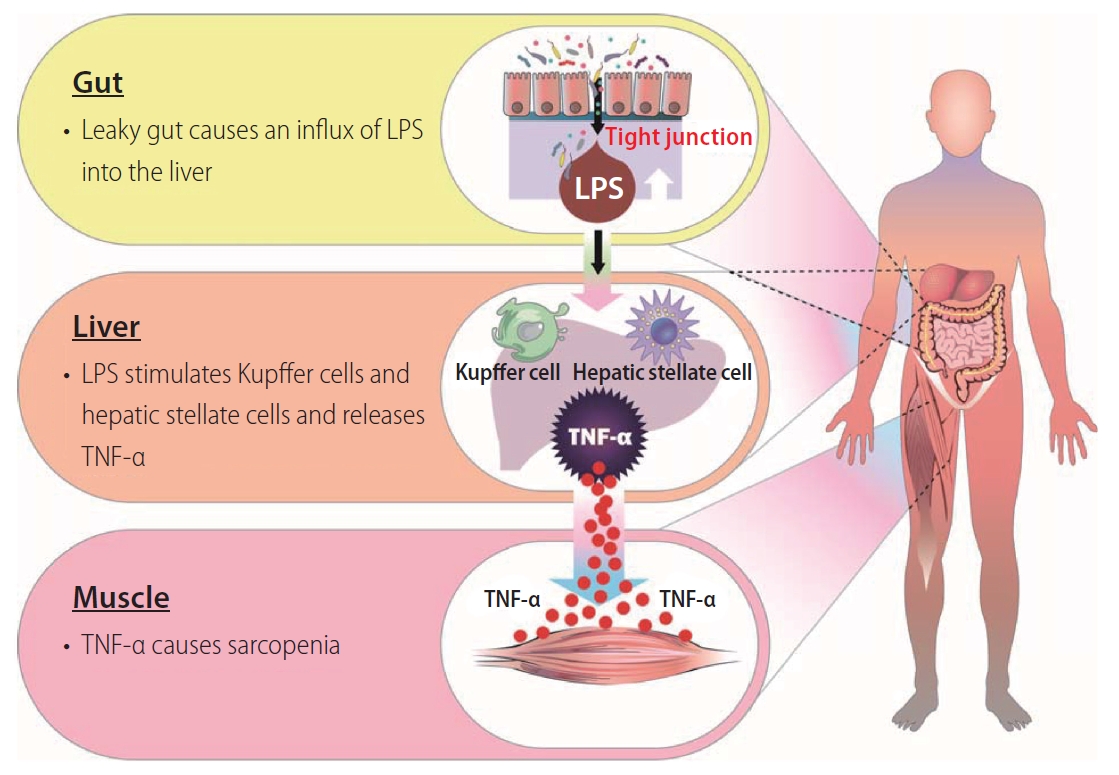
A proposed gut-liver-muscle axis of liver cirrhosis-related sarcopenia. Disruption of the intestinal tight junction causes an influx of lipopolysaccharide into the liver. Lipopolysaccharide stimulates Kupffer cells and hepatic stellate cells and releases TNF-α. Then, TNF-α causes sarcopenia. LPS, lipopolysaccharide; TNF-α, tumor necrosis factor-α.
Hyperammonemia is also a risk factor for sarcopenia in patients with liver cirrhosis [17]. Rifaximin suppresses ammonia-producing colonic bacteria and improves hyperammonemia [18]. In addition, rifaximin alters the gut microbiome composition (Lactobacillus, Streptococcus, Veillonella), which contributes to reducing hyperammonemia and endotoxemia in cirrhosis [18]. Furthermore, rifaximin has been reported to increase circulating saturated and unsaturated fatty acids and to modulate the metabolism of the host [19,20]. Ammonia-lowering treatment, including rifaximin, has been reported to reverse sarcopenia in a rat model of hyperammonemia by restoring skeletal muscle proteostasis [21]. Han et al. [6] demonstrated that treatment with rifaximin increased muscle mass and myofiber diameter in a rat model of cirrhosis. However, no reduction in blood ammonia levels was observed in rifaximin-treated rats compared to control rats. In contrast, rifaximin significantly reduced serum TNF-α levels and muscular expression of myostatin. Rifaximin has been reported to upregulate ZO-1 and reduce portal endotoxin levels in a rat model of liver cirrhosis [22]. Rifaximin has also been reported to reduce endotoxin activity and improve intestinal permeability, as evaluated by serum soluble CD163 and mannose receptors in patients with liver cirrhosis [23]. Accordingly, rifaximin may tighten the intestinal barrier and suppress serum TNF-α levels, leading to an improvement in sarcopenia with downregulation of myostatin expression.
The study by Han et al. showed that TNF-α is involved in the pathogenesis of sarcopenia in a rat model of liver cirrhosis. They also showed that rifaximin reduced serum TNF-α levels and improved sarcopenia in a rat model of liver cirrhosis. However, this study had some limitations. First, the pathogenesis of an increase in intestinal permeability remains unclear. Rifaximin is a non-systemic antibiotic that has been reported to alter the gut microbiota components in patients with liver cirrhosis [18]. Gut microbiota components are associated with various metabolites that regulate intestinal permeability and inflammatory cytokines [11,24]. Therefore, it is important to evaluate the impact of alterations in gut microbiota components and their metabolites on intestinal permeability. Second, it remains unclear whether rifaximin has an additive effect on nutritional and exercise therapies for sarcopenia. Third, it also remains unclear whether improvement of sarcopenia suppresses disease progression, development of life-threatening complications, and mortality in patients with liver cirrhosis. Further studies should focus on the effects of the combination of nutritional/exercise therapies and rifaximin treatment on long-term outcomes in patients with liver cirrhosis.
Alterations in intestinal permeability and inflammatory cytokines are crucial in the pathogenesis of sarcopenia in patients with liver cirrhosis. Further elucidation of the gut-liver-muscle axis may serve as a therapeutic strategy for sarcopenia.
Notes
Authors’ contributions
All authors were responsible for the interpretation of data, the drafting, and the critical revision of the manuscript for important intellectual content.
Conflicts of Interest
Takumi Kawaguchi received lecture fees from Janssen Pharmaceutical K.K., Mitsubishi Tanabe Pharma Corporation, and Otsuka Pharmaceutical Co., Ltd. The other author has no conflicts of interest.
Acknowledgements
This work was supported by Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research (C) JP20K08395 and by the Research Program on Hepatitis from Japan Agency for Medical Research and Development, AMED under 21fk0210094.
Abbreviations
TNF-α
tumor necrosis factor-α
ZO-1
zonula occludens-1