RNA interference as a novel treatment strategy for chronic hepatitis B infection
Article information

Abstract
Chronic hepatitis B (CHB) is a major cause of liver-related morbidity and mortality. Functional cure of CHB, defined as sustainable hepatitis B surface antigen (HBsAg) seroclearance, is associated with improved clinical outcomes. However, functional cure is rarely attainable by current treatment modalities. RNA interference (RNAi) by small-interfering RNA (siRNA) and anti-sense oligonucleotide (ASO) has been studied as a novel treatment strategy for CHB. RNAi targets post-transcriptional messenger RNAs and pregenomic RNAs to reduce hepatitis B virus (HBV) antigen production and viral replication. By reducing viral antigens, host immune reconstitution against HBV may also be attained. Phase I/II trials on siRNAs have demonstrated them to be safe and well-tolerated. siRNA is effective when given in monthly doses with different total number of doses according to different trial design, and can lead to sustainable dose-dependent mean HBsAg reduction by 2–2.5 log. Incidences of HBsAg seroclearance after siRNA therapy have also been reported. ASOs have also been studied in early phase trials, and a phase Ib study using frequent dosing regimen within 4 weeks could achieve similar HBsAg reduction of 2 log from baseline. Given the established efficacy and safety of nucleos(t) ide analogues (NAs), future RNAi regimens will likely include NA backbone. While the current evidence on RNAi appears promising, it remains undetermined whether the potent HBsAg reduction by RNAi can result in a high rate of HBsAg seroclearance with durability. Data on RNAi from phase IIb/III trials are keenly anticipated.
BACKGROUND
Chronic hepatitis B (CHB) affects over 290 million patients worldwide and is a major cause of liver-related morbidity and mortality [1]. Established treatment modalities for CHB include nucleos(t)ide analogues (NAs) and pegylated interferon. While current agents are effective in suppressing the viral load, complete cure and sterilization of hepatitis B virus (HBV) are not achievable due to persistence of covalently closed circular DNA (cccDNA) and viral integration into the host genome [2]. Chronic exposure to high levels of HBV antigens also lead to host immune dysfunction/anergy and result in inability of the immune system to clear the virus [2]. Functional cure, defined as sustained loss of hepatitis B surface antigen (HBsAg), is a favorable endpoint in CHB due to its association with fibrosis regression [3] and reduced hepatocellular carcinoma risk [4]. It is also regarded as a clinical indicator of adequate host immune control on HBV [5].
Multiple novel therapeutics, broadly classified as virus-targeting agents and immunomodulators, are being actively investigated as strategies for achieving functional cure [5]. Virus-targeting agents inhibit different steps of the HBV lifecycle and agents in development include: 1) entry inhibitors: which inhibit HBV docking and hepatocyte entry [6]; 2) polymerase inhibitors: which inhibit HBV polymerase competitively (conventional NAs) or non-competitively (novel active site polymerase inhibitor nucleotide) [7]; 3) RNA silencers: which inhibit HBV messenger RNA (mRNA) translation and viral protein production [8]; 4) capsid assembly modulators: which inhibit HBV nucleocapsid assembly and pregenomic RNA encapsidation [9]; 5) viral protein export inhibitors: which inhibit HBV antigen release from hepatocytes [10,11]; and 6) farnesoid X receptor (FXR) agonist: which reverses the interaction between FXR and the HBV X protein to interfere with transcription regulation [12]. Immunomodulators utilize an alternative strategy which is different from virus-targeting agents. Immunomodulators reconstitute the patient’s immune system to induce responses against HBV. Immunomodulators that have been studied include toll-like receptor agonists [13], immune checkpoint inhibitors [14], T-cell modulators [15], therapeutic vaccines [16], and recombinant immunoglobulins [17].
Among the above therapeutic means, RNA interference (RNAi) was one of the earliest agents developed and has demonstrated promising results [5]. RNAi involves the use of homologous nucleotide strands to target post-transcriptional HBV mRNA, which in turn inhibit downstream viral protein production [5]. RNAi can suppress HBsAg to low levels, which is an important surrogate outcome that predicts HBsAg seroclearance [18]. With HBsAg reduction through RNAi, T-cell anergy may be reversed and reconstitution of the host immune response may be achievable. This article will review the mechanisms and evidence on using RNAi in CHB.
RNAi
Mechanism of RNAi
In 1998, Fire et al. [19] first reported non-coding double-stranded RNA to have potent and specific effects on posttranscriptional gene silencing in Caenorhabditis elegans (roundworm). The non-coding double-stranded RNA was named small-interfering RNA (siRNA) and this phenomenon was termed RNAi [19].
siRNA has a passenger strand (sense) and guide strand (antisense), with the guide strand being complementary to target mRNA. siRNA is taken into the cytoplasm via endocytosis, after which it interacts with Dicer (RNase III endonuclease), Argonaute (RNase) and transactivation response element RNA-binding protein (RNA-binding cofactor) to form the RNA-induced silencing complex loading complex (RLC) [20]. The RLC retains the siRNA guide strand and removes the passenger strand to form a mature RNA-induced silencing complex (RISC). RISC can subsequently bind to target mRNA that has complementary sequence to the siRNA guide strand [21].
After binding, RISC induces gene silencing through a variety of mechanisms, which may vary between organisms. Argonaute-induced mRNA degradation is the most well-described, where Argonaute cleave the target mRNA between nucleotides 10 and 11, inducing exonuclease degradation of the cleaved oligonucleotides [22]. RISC can also directly inhibit RNA translation through deadenylation of the poly(A) tail of mRNA, blocking protein interactions between initiation factors, and inducing premature termination of translation [23,24]. Finally, RISC can induce formation of heterochromatin in the target DNA through histone methyltransferases to induce epigenetic changes [25]. RISC is a multiple turnover enzyme, hence a single siRNA can silent multiple mRNA transcripts after activation into RISC [26]. Figure 1 depicts the mechanism of RNAi.
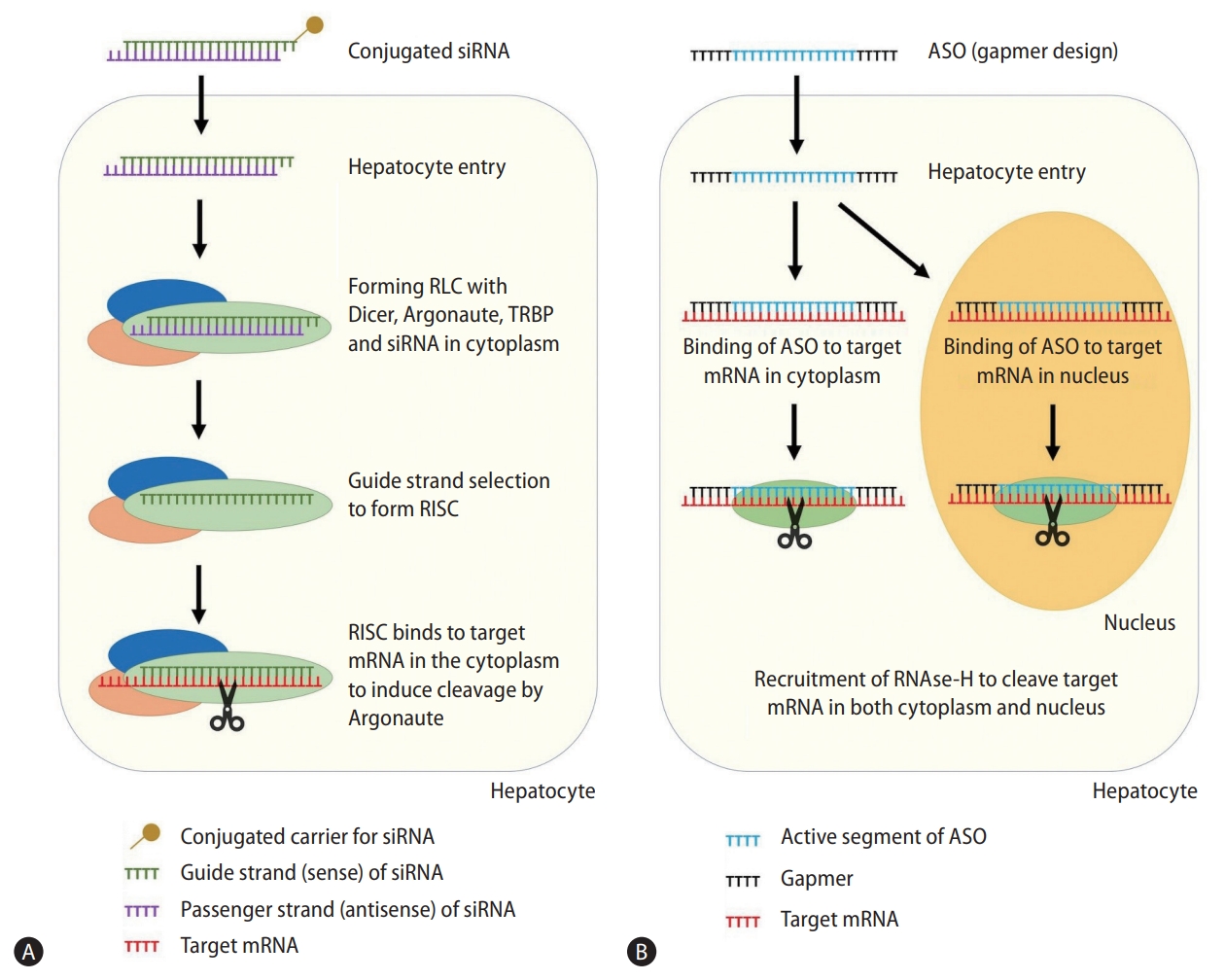
Mechanism of small-interfering RNA (A) and antisense oligonucleotides (B). siRNA, small-interfering RNA; RLC, RNA-induced silencing complex loading complex; TRBP, transactivation response element RNA-binding protein; RISC, RNA-induced silencing complex; mRNA, messenger RNA; ASO, antisense oligonucleotide.
RNAi as a therapeutic strategy for viral infections
RNAi is a versatile technique that can target any gene with an identifiable sequence, overcoming the challenge of selecting downstream “druggable” targets. Patisiran, an siRNA targeting hereditary transthyretin amyloidosis, became the first siRNA therapeutic approved by the US Food and Drug Administration in 2018 [27]. Since then, the field of siRNA therapeutics has been rapidly expanding. Due to the versatility of siRNA, its use is currently studied in a wide range of diseases including viral infections, genetic conditions, heart failure, chronic kidney disease, and malignancies [28]. As a drug class, siRNAs have also demonstrated impressive safety data and are generally well-tolerated [28].
At present, siRNA is studied in chronic viral infections that cannot be eliminated by current therapeutics, such as CHB [8] and human immunodeficiency virus (HIV) infection [29,30]. siRNA has also been studied in viruses that do not have effective treatment available, such as respiratory syncytial virus [31], poliovirus [32], and Ebola virus [33].
A key consideration in developing siRNA antivirals involves appropriate sequence selection. The selected RNA sequence should be highly specific to conserved sequences in the targeted viral genome, such that pan-genotypic antiviral effects can be exerted [34]. Specific siRNA sequences may also reduce off-target effects on the host genome that induce undesirable drug toxicity [35]. The optimal length of siRNA is 21 nucleotides with two nucleotides overhanging on the 3’ end, as longer sequences increase the risk of off-target effects [36]. Advanced bioinformatics techniques and specialized software are utilized for filtering inappropriate sequences and predicting effective sequences [37].
Structural optimization is critical for siRNA antivirals. Due to naturally occurring nucleases, unmodified siRNA is rapidly broken down in human serum [38]. Furthermore, due to the presence of a phosphate backbone and anionic charge, unmodified siRNA is hydrophilic and cannot diffuse through negatively charged cell membranes [39]. Finally, siRNA has immune stimulatory effects and can induce unwanted nonspecific interferon responses through double-stranded RNA-dependent protein kinase [40] and toll-like receptors [41]. Chemical modification of the siRNA phosphate backbone can tackle all three challenges of siRNA instability, cellular entry, and inadvertent immune activation. By replacing the 2’-OH group by 2’-O-methyl or 2’-F-nucleotide on the phosphate backbone, siRNA can be protected from serum nucleases [42], has reduced off-target effects [43], has minimal unwanted immune stimulatory responses [44], and at the same time, has increased potency by 500-fold [45].
Delivery systems are required to carry siRNA into target cells and both viral and non-viral vectors have been studied. Adenovirus vector is the most commonly used viral vector, as it is non-toxic, easy to produce, and adequate experience is available from its use in vaccines [46]. Non-viral vectors, on the other hand, include polymers, aptamers, peptides, liposomes, antibodies and lipid nanoparticles [47]. The choice of delivery system depends on the desired target of the siRNA. For example, N-acetylgalactosamine (GalNAc) is ideal for liver-directing siRNA as GalNAc binds to Asialoglycoprotein receptors, a specific and abundant receptor in hepatocytes [48]. GalNAc-conjugation minimizes systemic siRNA exposure, reduces infusion reactions, lengthens dosing intervals, and enables administration through subcutaneous infusion [48].
Designing RNAi therapeutics for HBV
HBV is characterized by its four open reading frames (ORFs) that encode for hepatitis B precore/core, polymerase, surface, and X proteins respectively. The four ORFs have overlapping sequences and have the same polyadenylation signal at the 3’ end in the core protein coding region. This enables interference of all four HBV transcripts with a single siRNA, reducing all downstream viral protein and pregenomic RNA production. Viral replication will therefore be reduced by siRNA, which may indirectly reduce the cccDNA reservoir [49]. siRNA is hence able to directly or indirectly interfere with multiple steps of the viral lifecycle. Figure 2 depicts the mechanism of RNAi against HBV.

Mechanism of RNA interference as a treatment strategy in chronic hepatitis B. HBV, hepatitis B virus; cccDNA, covalently closed circular DNA; pgRNA, pregenomic RNA; mRNA, messenger RNA; siRNA, small-interfering RNA; ASO, antisense oligonucleotide.
Viral mutation is a concern in designing siRNA against HBV, as a single base mismatch could lead to loss of siRNA effect. Rapid emergence of mutant strains after siRNA treatment has been reported in RNA viruses including HIV [50] and hepatitis C virus [51]. HBV, while being a DNA virus, requires reverse transcriptase for replication. As reverse transcriptase does not have proofreading capability, HBV has a mutation rate of 2×104 base substitutions/site/year, which is 100 times higher than other DNA viruses [52]. Nonetheless, the mutation rate of HBV is still 100–1,000 times lower than that of RNA viruses [52]. siRNA-induced mutants have not been reported in HBV, yet siRNA can exert selection pressure on preexisting resistant HBV quasispecies [53]. Targeting functionally vital and conserved regions [53] or using multiple siRNA triggers [54] are essential in minimizing the potential impact of HBV mutants in siRNA therapy.
siRNA may also restore the host immune response against HBV. A major contributor in the chronic persistence of HBV is host immune suppression after long term exposure to high levels of immune repressive viral antigens, particularly HBsAg [32]. CHB is associated with impairment of host immune pathways including Toll-like receptor signaling [55], immune checkpoint signalling [56], and increased immunosuppressive T-regulatory cells activity [57]. These actions ultimately lead to reduction and dysfunction of HBV-specific T-cell clones [58,59], resulting in numerical and functional T-cell deficits against HBV. Through its potent effect in reducing HBV viral antigens, siRNA may indirectly reconstitute the immune system. On one hand, long term viral suppression with NAs can only minimally reduce viral antigen levels as they inhibit the reverse transcriptase but cannot directly target HBV cccDNA [60,61]. siRNA on the other hand, is predictably associated with more pronounced HBsAg reduction.
Antisense oligonucleotides (ASOs), an alternative form of gene silencing
Antisense oligonucleotide (ASO) is a related yet distinct drug class from siRNA. Like siRNA, ASO utilizes complementary nucleotide binding to exert post-transcriptional gene silencing on target mRNA (Fig. 1).
Key differences should be noted between siRNA and ASO (Table 1). First, siRNAs are double-stranded RNA with ideal length of 21 nucleotides plus two 3’ end overhanging nucleotides. In contrast, ASOs are single-stranded DNA of 15 to 25 nucleotides in length [62]. ASOs are designed as gapmers, with a central unmodified complementary DNA sequence flanked by modified RNA-like segments on both sides. Gapmer structure enhances the affinity of ASO to its target sequence and increases its resistance to nuclease degradation [63].
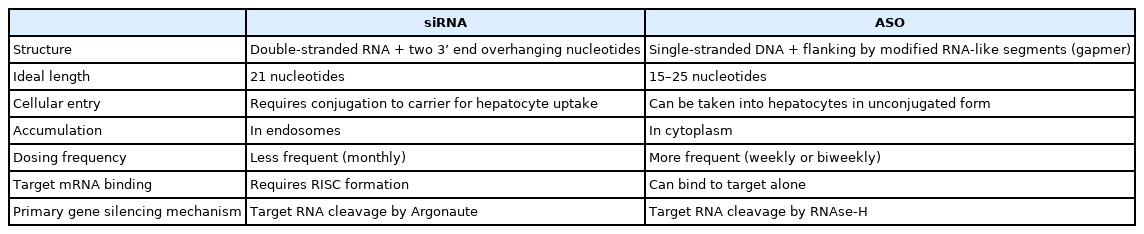
Differences between small-interfering RNA and antisense oligonucleotides
Secondly, unmodified siRNA requires carriers for cellular entry, whereas unconjugated ASO is taken up into hepatocytes through receptor mediated pathways [64]. After cellular entry, siRNA accumulates in endosomes and requires less frequent dosing [65,66], whereas ASO accumulates in cytoplasm and requires more frequent dosing [67]. While conjugation to carriers is not a necessity for ASO, GalNAc-conjugation in ASO can enhance hepatocyte uptake and reduce systemic exposure, similar as the effect of GalNAc-conjugation in siRNA [68].
Thirdly, siRNA requires RISC formation for target mRNA binding, as passenger strand removal is integral in the exposure of complementary nucleotides on the guide strand [21], whereas ASO can bind to target mRNA alone.
Fourthly, siRNA and ASO have distinct mechanisms of gene silencing. siRNA mediates its action through RISC as described above. In contrast, ASO most commonly mediates its action through recruitment of RNase-H (a family of endonucleases) in both the cytoplasm and nucleus to cleave target RNA [69]. ASO may also inhibit RNA translation through inhibiting 5’ cap formation [70] or blocking ribosomal subunit attachment [71].
ASO has been studied as a novel therapeutic in CHB [72], and the evidence on ASO will be discussed below.
PRECLINICAL AND CLINICAL EVIDENCE IN CHB
Wooddell et al. [73] first reported the efficacy of siRNA in suppressing HBV in animal models. A single dose of ARC-520 induced 3.0 log IU/mL HBsAg reduction in mice [73] and 2.7 log IU/mL HBsAg reduction in hepatitis B e-antigen (HBeAg)-positive chimpanzees [74]. However, HBsAg reduction was comparatively lower in HBeAg-negative chimpanzees (0.9 log IU/mL). The discrepancy in HBsAg responses between HBeAg-positive and negative subjects was an unexpected finding. The investigators proceeded with a range of genome and animal experiments, which demonstrated that HBsAg was not only expressed from HBV cccDNA, but can also be produced from integrated HBV DNA. As ARC-520 was designed to target sequences between DR1 and DR2 regions of HBV (sequences commonly lost during HBV integration), it was unable to target HBV integrants, leading to differential responses in HBeAg-positive and negative subjects [74]. Newer generation siRNA is designed to target transcripts upstream of integration hotspots [75] or have multiple triggers [76,77], enabling targeting of all HBV transcripts from cccDNA and HBV integrants.
After ARC-520, a range of newer RNA silencers have demonstrated promising data in in-vitro and in-vivo studies [78-80], and they have already entered clinical trials (Table 2). HBsAg suppression is the key outcome measured in the trials and the results are summarized below (Table 3).
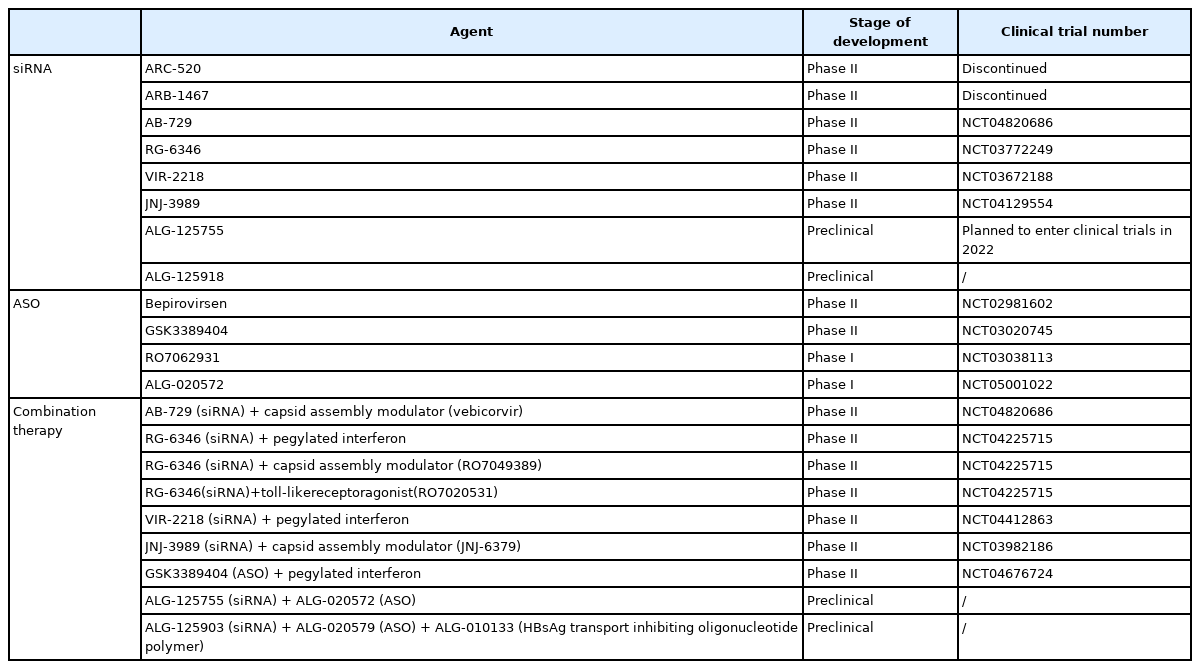
RNA silencers in development

Effects of small-interfering RNA on hepatitis B surface antigen in clinical trials
ARC-520
As already described above, ARC-520 is the first-in-class siRNA for CHB. It contains two siRNA triggers targeting overlapping regions in the X ORF of HBV cccDNA. It is conjugated to cholesterol and is administered intravenously. Early safety studies demonstrated that adverse events were comparable in ARC-520 and placebo groups, with no serious adverse events noted [81].
The Heparc-2001 trial was a phase II trial that tested the efficacy of a single dose of ARC-520 in CHB patients. Four arms of the trial consisted of HBeAg-negative NA treated patients who received ARC-520 at doses ranging from 1–4 mg/kg. Serum HBsAg declined within the first eight days of ARC-520 dosing. HBsAg responses correlated with ARC-520 dosage and the 4 mg/kg group had HBsAg reduction by 0.3 log IU/mL [74]. In a separate arm of treatment-naïve patients (six HBeAg-positive and six HBeAg-negative), a single dose of 4 mg/kg ARC-520 was started concurrently with entecavir. Excluding one HBeAg-positive patient who had low baseline HBeAg level and was considered to be transitioning to HBeAg-negative status, the HBeAg-positive group had HBsAg reduction by 1.4 log IU/mL. In contrast, HBeAg-negative treatment-naïve patients did not have significant HBsAg reduction [74]. As discussed above, this differential response in HBeAg-positive and negative patients is attributable to the inability of ARC520 in targeting HBV integrants.
In the Heparc-2002 and Heparc-2003 trials, patients on NAs were randomized to receive four monthly doses of 2 mg/kg ARC-520 or placebo. ARC-520 led to HBsAg reduction by 0.54 log IU/mL and 0.38 log IU/mL in HBeAg-positive and negative patients respectively. The HBsAg response was sustainable for over 85 days post-treatment [82].
Eight patients (three HBeAg positive and five HBeAg negative) from the Heparc-2001 trial were recruited to an extension trial. The patients received up to nine monthly doses of 4 mg/kg ARC-520 around 8 to 10 months after initial dosing and were followed up for up to 33 months afterwards. Among HBeAg-positive patients, one patient achieved HBsAg seroclearance, whereas the other two patients had HBsAg reduction of 1.7 log IU/mL and 3.5 log IU/mL from baseline respectively. Among HBeAg-negative patients, one patient achieved HBsAg seroclearance, whereas the other patients had a mean HBsAg reduction of 0.4 log IU/mL from baseline. The HBsAg reduction was sustainable for up to 33 months after end of treatment [83].
The development of ARC-520 was terminated in November 2016, as incidences of mortality induced by the excipient of ARC-520 (not the active siRNA component) in non-human primates were reported.
ARB-1467
ARB-1467 has three siRNA triggers targeting the HBV S and X ORFs. It is delivered via lipid nanoparticles and is administered intravenously. In a 12-week trial on NA treated patients, 0.2 mg/kg or 0.4 mg/kg ARB-1467 was administered monthly for three doses, and mean HBsAg reduction of 0.6 to 0.9 log IU/mL was achieved. HBsAg reduction was sustained at end of follow-up (12 weeks). In the trial arms receiving 0.4 mg/kg ARB-1467, 45.5% of patients (five out of 11) achieved greater than 1 log IU/mL HBsAg reduction. ARB-1467 was well-tolerated, with mild injection reactions being the most commonly reported adverse event [76].
Due to the modest effect of monthly ARB-1467 on HBsAg decline, biweekly dosing of ARB-1467 was tested. In a second part of the ARB-1467 trial, 12 NA treated patients received five doses of biweekly ARB-1467. When compared with monthly dosing, patients on biweekly dosing had a greater magnitude of HBsAg reduction, achieving maximum individual HBsAg decline of 2.7 log IU/mL. HBsAg reduction was sustainable at 10 weeks [84].
The development of ARB-1467 was discontinued in March 2019, as the sponsor aimed to shift their focus to their newer siRNA product AB-729 [85].
AB-729
AB-729 is a subcutaneous GalNAc-conjugated siRNA with a single trigger targeting the HBV X protein. A phase II multiarm trial assessed the effects of AB-729 at different doses (60 mg or 90 mg) and dosing intervals (4-weekly, 8-weekly, or 12-weekly dosing for 24 weeks, followed by extension of dosing every 8 or 12 weeks) in NA treated patients. HBsAg reduction ranged from 1.86 to 2.16 log IU/mL in different trial arms, and the HBsAg decline was sustained up to 48 weeks [86]. Across the treatment arms, over 70% of patients achieved HBsAg below 100 IU/mL. HBsAg reduction also correlated with reduction of circulating HBV RNA and HBsAg isoforms [87], and was associated with an increase in HBV-specific T-cell activation markers [88]. Patients on AB-729 had an increase in interferon-gamma producing HBV specific T-cells and had transient alanine aminotransferase (ALT) flares that correlated with HBsAg decline. These findings support the role of AB-729 in inducing immune reconstitution against HBV [87,88]. Injection site reactions were common in AB-729, yet no serious adverse events, trial discontinuations or deaths were reported.
RG-6346
RG-6346 is a subcutaneous GalNAc-conjugated siRNA with a single trigger targeting the HBV S ORF. In a phase IIa trial, NA treated patients received four monthly doses of RG-6346. The mean HBsAg reduction were 1.64 log IU/mL, 1.91 log IU/mL and 1.87 log IU/mL in the 1.5 mg/kg, 3.0 mg/kg or 6.0 mg/kg RG-6346 arms respectively. Across treatment arms, 92% of patients achieved more than 1 log IU/mL HBsAg reduction and 58% of patients achieved HBsAg below 100 IU/mL. An additional NA naïve arm achieved HBsAg reduction of 1.02 log IU/mL with 3.0 mg/kg RG-6346. The HBsAg suppression was sustained for up to 64 weeks. RG-6346 was associated with self-resolving ALT flares that corresponded with declining HBsAg, suggesting treatment-induced immune reconstitution. Injection site reactions and flu-like symptoms were common adverse events in RG-6346 [89].
VIR-2218
VIR-2218 is a subcutaneous GalNAc-conjugated siRNA with a single trigger targeting the HBV X ORF. In a 48-week trial on NA treated patients, two monthly doses of VIR-2218 in the doses of 20 mg, 50 mg, 100 mg, or 200 mg were given. Across the treatment arms, 70.8% of patients had more than 1 log IU/mL HBsAg reduction. Among patients who achieved 1 log IU/mL HBsAg reduction, 70.6% of patients achieved HBsAg below 100 IU/mL. Suppression of HBsAg below 100 IU/mL was not sustainable at 48 weeks in the 20 mg and 50 mg VIR-2218 groups, whereas the response was sustained in 36.4% of patients in the 100 mg and 200 mg VIR-2218 groups. VIR-2218 had a favorable safety profile, with headache being its most common side effect [75].
JNJ-3989
JNJ-3989 is a subcutaneous GalNAc-conjugated siRNA with two triggers targeting the HBV X and S regions. In a phase II trial that recruited both NA treated and treatment-naïve patients, 3 monthly doses of JNJ-3989 at doses ranging from 100 mg to 400 mg were given, with NAs given concurrently with JNJ-3989 in the treatment-naïve group. Across the treatment arms, a mean HBsAg reduction by 1.93 log IU/mL along with decline in HBeAg and HBV RNA was documented. 97.5% of patients across the treatment arms achieved more than 1 log IU/mL HBsAg reduction, and the changes were sustained at 24 weeks [77].
Agents in preclinical phase
ALG-125918 is an siRNA designed with a novel 5’ cap phosphate mimic that enhances siRNA loading and cleavage efficiency. A single dose of 5 mg/kg ALG-125918 led to maximum HBsAg reduction by 1.5 log IU/mL in mice, which was sustainable for 90 days [90]. The development of ALG-125918 is ongoing.
ALG-125755 is a subcutaneous GalNAc-conjugated siRNA with a single trigger targeting the S ORF. A single dose of 5 mg/kg ALG-125755 led to HBsAg reduction by 1.5 log IU/mL in mice, and the effect was sustained at 6 weeks [91]. ALG-125755 will enter clinical trials in early 2022 [92].
Antisense oligonucleotides
This section will summarize the clinical evidence on ASO (Table 4).
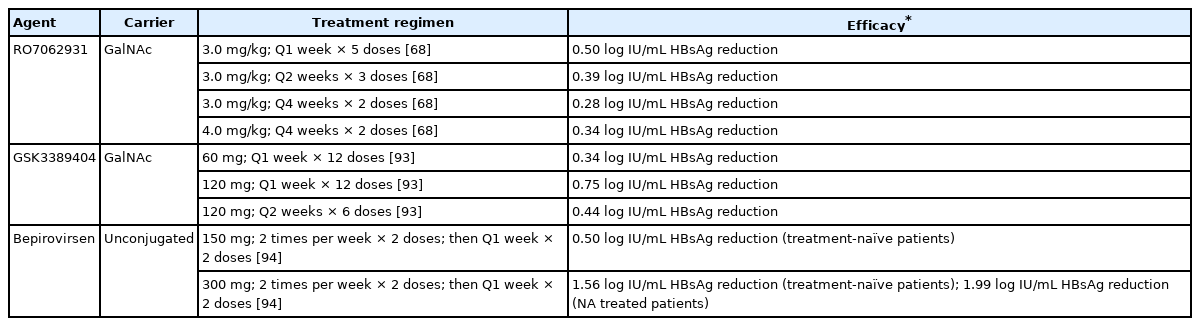
Effects of antisense oligonucleotides on hepatitis B surface antigen in clinical trials
RO7062931 is a subcutaneous GalNAc-conjugated gapmer ASO targeting a highly conserved sequence in the shared 3’ region. In a phase I trial, NA treated CHB patients received placebo or 2 monthly doses of RO7062931 at 0.5 mg/kg, 1.5 mg/kg or 3.0 mg/kg. A second part of the trial recruited patients to receive RO7062931 at 3.0 mg/kg biweekly for three doses, 3.0 mg/kg weekly for five doses or 4.0 mg/kg weekly for four doses. All patients dosed with 0.5 mg/kg to 3.0 mg/kg doses had dose-dependent and time-dependent HBsAg reduction, although dose-dependence was not observed in the 4.0 mg/kg arm, possibly due to small sample size. The greatest HBsAg decline of 0.50 log IU/mL was observed in the 3.0 m/kg weekly arm. All patients in treatment arms had HBsAg nadir 2 weeks after starting treatment, with levels returning to baseline within 12 weeks post-treatment. RO7062931 was safe and well-tolerated [68]. The development of RO7062931 was halted due to suboptimal magnitude of HBsAg reduction despite established target engagement.
GSK3389404 is a subcutaneous GalNAc-conjugated 2’-O-methoxyethyl gapmer ASO targeting the HBV X ORF. In a phase II trial, NA treated patients were given GSK3389404 at 60 mg weekly, 120 mg biweekly or 120 mg weekly for 12 weeks. HBsAg reduction was dose-dependent, with HBsAg decline of 0.34 log IU/mL, 0.44 log IU/mL, and 0.75 log IU/mL in the 60 mg weekly, 120 mg biweekly group, and 120 mg weekly groups respectively. HBsAg levels returned to baseline within 12 weeks post-treatment [93]. Similar to RO7062931, the modest HBsAg reduction effects precluded future development of GSK3389404.
Bepirovirsen (GSK3228836) is the unconjugated version of GSK3389404 that can be administered subcutaneously. A phase II trial recruited 24 treatment-naïve patients to have placebo, 150 mg bepirovirsen or 300 mg bepirovirsen; with initiation of NA after the first month. The trial also recruited seven NA treated patients to receive placebo or 300 mg bepirovirsen. Bepirovirsen was administered across four weeks (twice weekly for 2 weeks, then weekly for 2 weeks), with subsequent follow-up for 26 weeks. In the treatmentnaïve group, 150 mg bepirovirsen and 300 mg bepirovirsen led to end-treatment mean HBsAg reduction by 0.50 log IU/mL and 1.56 log IU/mL, respectively. In the treatment-experienced 300 mg bepirovirsen group, the mean HBsAg reduction was 1.99 log IU/mL. Two treatment-naïve patients and two treatment-experienced patients, all given 300 mg bepirovirsen, achieved transient unquantifiable HBsAg, with the durability of unquantifiable HBsAg lasting for 57 to 126 days after treatment initiation. Bepirovirsen was well-tolerated. Most treatment-emergent adverse events were mild injection site reactions. Transient self-resolving ALT flares which coincided with HBsAg reduction were noted in the treatment arms, suggesting immune clearance of infected hepatocytes [94].
ALG-020572, a GalNAc-conjugated ASO targeting the S ORF, demonstrated rapid absorption and distribution in mice. After administration of ALG-020572 in 3-day intervals in mice for seven doses, dose-dependent HBsAg reduction was demonstrated, with a maximum of 1.1 log IU/mL HBsAg reduction in the 10 mg/kg ALG-020572 arm. Pharmacokinetic studies in non-human primates demonstrated ALG-020572 to have an intrahepatic half-life of 12 days, supporting a weekly regimen in humans. ALG-020572 is currently evaluated in phase I clinical trials.
Combination therapy
Combination regimens involving RNAi have also been studied, as RNAi alone have not demonstrated consistent HBsAg seroclearance effects in current trials. Combination regimens aim to target different steps of the HBV lifecycle to lead to synergistic antiviral effects.
VIR-2218 has been tested in combination with pegylated interferon alpha-2a. The combination trial recruited NA treated patients to receive 6 monthly doses of 200 mg VIR-2218 along with varying regimens of pegylated interferon alpha-2a in different trial arms. Ninety-two percent of patients in all VIR-2218 arms (with or without interferon) achieved HBsAg below 100 IU/mL by week 24. Concurrent initiation of VIR-2218 and interferon achieved the highest HBsAg reduction by 2.55 log IU/mL at end of treatment, whereas HBsAg decline was relatively less pronounced in patients with VIR-2218 monotherapy (1.89 log IU/mL) or with add-on interferon after VIR-2218 (2.03 log IU/mL). Among the 37 patients on combination therapy, three patients (8.1%) had unquantifiable HBsAg at week 24. Only data up till end of treatment (24 weeks) were presented in abstract form in 2021 and the complete results including post-treatment HBsAg sustainability are pending [95]. Higher incidences of injection site reactions and flu-like symptoms were documented in interferon-containing arms. One patient discontinued from the trial due to interferon-related depression and the patient recovered after drug discontinuation [95].
The REEF-1 trial is a 48-week trial which recruited NA treated patients to test the combination of JNJ-3989 with the capsid assembly modulator JNJ-6379. Patients in the REEF-1 trial were randomized to receive capsid assembly modulator (250 mg JNJ-6379) monotherapy, JNJ-3989 monotherapy at 40 mg/100 mg/200 mg, or combination of 100 mg JNJ-3989 plus capsid assembly modulator (250 mg JNJ-6379). JNJ-3989 was administered at monthly doses for 48 weeks. The capsid assembly modulator was given daily for 48 weeks. HBsAg decline was dependent on dosage of JNJ-3989. In the 200 mg JNJ-3989 group, 74.7% of patients achieved HBsAg below 100 IU/mL, and the group had mean HBsAg reduction of 2.6 log IU/mL. Capsid assembly modulator monotherapy only led to HBsAg decline by 0.07 log IU/mL, whereas the combination group (100mg JNJ-3989 plus capsid assembly modulator) achieved 1.8 log IU/mL HBsAg decline at end of treatment; both of which were lower than the levels achieved by the 200 mg JNJ-3989 group [96]. At 24 weeks after the end of treatment, the HBsAg levels had a gradual increasing trend towards baseline, however the 200 mg JNJ-3989 group still had mean HBsAg 1.9 log IU/mL lower than baseline. Among patients in the REEF-1 trial, flu-like symptoms were the most common adverse events reported. Five patients were discontinued from the trial, as three patients in capsid assembly modulator containing arms had renal derangement, one patient in the 40 mg JNJ-3989 arm had virologic breakthrough, and one patient in the 40 mg JNJ-3989 arm had insomnia [96].
Novel combination regimens including ALG-125755 (siRNA) + ALG-020572 (ASO) [97], and ALG-125903 (siRNA) + ALG-020579 (ASO) + ALG-010133 (HBsAg transport inhibiting oligonucleotide polymer) [98] have shown synergistic effects in in-vitro and in-vivo experiments, and the development of these novel regimens are ongoing. A range of combination trials including siRNA with toll-like receptor agonists, capsid assembly modulators or pegylated interferon are also ongoing and the results are keenly anticipated.
OVERVIEW OF CURRENT EVIDENCE AND CONCLUSION
Clinical trials have consistently demonstrated siRNA to be safe, with most adverse events being mild injection reactions or flu-like symptoms [76,89,94]. ALT flares can occur in siRNA therapy, but are usually transient and associated with HBsAg reduction, suggesting immune reconstitution and elimination of infected hepatocytes [87-89,94].
siRNA has demonstrated potent HBsAg reduction effects. Among the newer generation siRNA (excluding the older generation ARC-520 and ARB-1467), mean HBsAg suppression by 2–2.5 log is achievable, with over 90% of patients in high dose treatment arms reaching over 1 log IU/mL HBsAg reduction, and 50–97% of patients having HBsAg suppressed to below 100 IU/mL [75,77,86,89]. HBsAg reduction in siRNA was sustainable after end of treatment [75,86,89], and incidences of HBsAg seroclearance have been documented [83,95]. Nonetheless it remains unclear whether these potent HBsAg reduction effects can be translated into durable HBsAg seroclearance. Furthermore, larger scale studies comparing HBeAg-positive and -negative patients are required to determine whether the drug effects are generalizable to different CHB subgroups. The long term data from current trials on different CHB populations are keenly anticipated.
Multiple dose regimens are the standard for siRNA, as multiple dosing led to higher magnitude of HBsAg reduction and more sustainable effects than single dosing [83,84,86,89,95,96]. The current evidence supports monthly administration of siRNA, with AB-729 demonstrating potent effects even in 8 or 12-weekly dosing [86]. The total dosing duration for siRNA is less well-defined, as different treatment durations (ranging from 2 months to 1 year) were adopted in the current trials. The decision on ideal treatment durations will likely depend on the durability of post-treatment HBsAg suppression, and trials with longer follow-up will be required to monitor for HBsAg seroclearance. These issues have important implications for the future development of RNAi therapy.
Similar to siRNA, ASO is safe and well-tolerated [68,94]. While siRNA is administered monthly, ASO requires more frequent dosing (weekly or biweekly), reflecting the inherent pharmacokinetic discrepancy between siRNA and ASO. Bepirovirsen showed potent HBsAg reduction effects and even transiently suppressed HBsAg to unquantifiable levels in four patients. A potential explanation for this finding is high cumulative dosing, as six doses of 300 mg bepirovirsen were administered within one month in the trial, which was comparatively higher than the dosing in other ASO and siRNA trials [94].
Before deciding on the necessity of combining RNAi with other novel antivirals to achieve functional cure in all patients, we need to determine the probability of subsequent HBsAg seroclearance in patients who have achieved low HBsAg levels (e.g., <100 IU/mL) by RNAi. If the HBsAg seroclearance rate is reasonably high in a short period of time after stopping RNAi, we may not need to combine RNAi with other novel agents of different classes. Nevertheless, combination therapy may be necessary for patients who remain to have high HBsAg levels. With the established efficacy and safety of NAs, future RNAi treatment regimens will likely be combined with an NA backbone. Adding a third anti-HBV agent to RNAi and NA may lead to synergistic antiviral effects. As RNAi has potential effects of immune reconstitution, the addition of immunomodulators may further boost the host antiviral immune system. Indeed, combining VIR-2218 and pegylated interferon alpha-2a led to greater HBsAg suppression than either agent alone [95]. The current data suggest that combining siRNA with interferon can yield a synergistic effect. Nonetheless, other combination regimens of siRNA with immunomodulators including siRNA + therapeutic vaccines and siRNA + Toll-like receptor agonists are investigated in ongoing trials [99]. Combining siRNA with novel virus-targeting agents has also been investigated. Notably, preclinical experiments demonstrated synergistic effects when combining siRNA and ASO, two RNA silencers that share the same target (mRNA). Although the underlying mechanism and clinical efficacy of this combination is yet to be determined [97], it would be interesting to determine whether the synergistic effect is related to the full utilization of different RNA degradation pathways as described above. On the other hand, combination of siRNA and capsid assembly modulator has failed to yield synergistic effects in an ongoing trial [96]. This highlights the need for meticulous selection of combination agents with siRNA, and a range of combination trials involving agents with different mechanisms are ongoing.
To conclude, RNAi is a safe technique that can induce potent and sustainable HBsAg suppression. Research on RNAi is rapidly evolving and early phase data has been promising. Multiple trials are ongoing, and with further development, RNAi may emerge as a novel treatment strategy that shifts the paradigm of CHB therapy.
Notes
Authors’ contributions
RWHH was involved in data interpretation and drafting of the manuscript. LYM and WKS were involved in critical revision of the manuscript. MFY was involved in study concept, critical revision of the manuscript, and overall study supervision. All authors have seen and approved the final version of the manuscript.
Conflicts of Interest
MF Yuen is an advisory board member and/or received research funding from AbbVie, Arbutus Biopharma, Assembly Biosciences, Bristol Myer Squibb, Dicerna Pharmaceuticals, GlaxoSmithKline, Gilead Sciences, Janssen, Merck Sharp and Dohme, Clear B Therapeutics, Springbank Pharmaceuticals; and received research funding from Arrowhead Pharmaceuticals, Fujirebio Incorporation and Sysmex Corporation. WK Seto received speaker’s fees from AstraZeneca and Mylan, is an advisory board member of CSL Behring, is an advisory board member and received speaker’s fees from AbbVie, and is an advisory board member, received speaker’s fees and researching funding from Gilead Sciences. The remaining authors have no conflict of interests.
Abbreviations
ALT
alanine aminotransferase
ASOs
antisense oligonucleotides
CCCDNA
covalently closed circular DNA
CHB
chronic hepatitis B
FXR
farnesoid X receptor
GalNAc
N-acetylgalactosamine
HBeAg
hepatitis B e-antigen
HBsAg
hepatitis B surface antigen
HBV
hepatitis B virus
HIV
human immunodeficiency virus
mRNA
messenger RNA
NAs
nucleos(t)ide analogues
ORFs
open reading frames
RISC
RNA-induced silencing complex
RLC
RNA-induced silencing complex loading complex
RNAi
RNA interference
siRNA
small-interfering RNA