The emerging roles of extracellular vesicles as intercellular messengers in liver physiology and pathology
Article information

Abstract
Extracellular vesicles (EVs) are membrane-enclosed particles released from almost all cell types. EVs mediate intercellular communication by delivering their surface and luminal cargoes, including nucleic acids, proteins, and lipids, which reflect the pathophysiological conditions of their cellular origins. Hepatocytes and hepatic non-parenchymal cells utilize EVs to regulate a wide spectrum of biological events inside the liver and transfer them to distant organs through systemic circulation. The liver also receives EVs from multiple organs and integrates these extrahepatic signals that participate in pathophysiological processes. EVs have recently attracted growing attention for their crucial roles in maintaining and regulating hepatic homeostasis. This review summarizes the roles of EVs in intrahepatic and interorgan communications under different pathophysiological conditions of the liver, with a focus on chronic liver diseases including nonalcoholic steatohepatitis, alcoholic hepatitis, viral hepatitis, liver fibrosis, and hepatocellular carcinoma. This review also discusses recent progress for potential therapeutic applications of EVs by targeting or enhancing EV-mediated cellular communication for the treatment of liver diseases.
INTRODUCTION
Extracellular vesicles (EVs) are lipid bilayer-enclosed particles secreted from nearly all types of cells without replicable nuclei [1]. EVs can be classified by their processes of biogenesis (exosomes, microvesicles, and apoptotic bodies) or size, (small EVs <200 nm and medium/large EVs ≥200 nm) [1,2]. Exosomes are released by fusion of multi-vesicular bodies with the plasma membrane [3], while microvesicles are directly shed from the plasma membrane, and apoptotic bodies are produced by membrane blebbing of dying cells [4]. The release of EVs into the intercellular space mediates communication by either releasing EV cargo into tissue microenvironments or activating receptors on recipient cells via ligands on the EV surface. Alternatively, EVs can be endocytosed by specific recipient cells and release their cargo into the cytoplasm [5]. Once released, EVs mediate intercellular interactions for a wide range of physiological and pathological processes by transferring their cargo. EV-encapsulated cargoes are protected against enzymatic activities and can be shipped to distant organs or tissues to mediate interorgan crosstalk [6-8].
The liver is a dynamic organ that plays crucial roles to maintain body homeostasis, including gluconeogenesis, detoxification of xenobiotics, lipid metabolism, urea synthesis, and plasma protein synthesis [9]. However, the liver can be damaged by various external stimuli, including viral infection, alcohol consumption, and lipid overloading. Repeated and prolonged liver damage may lead to chronic liver diseases (CLDs) characterized by excessive steatosis, inflammation, and fibrosis, which are susceptible conditions for end-stage liver diseases such as cirrhosis and hepatocellular carcinoma (HCC) [10,11]. Intercellular communication is important for regulating these pathological processes of the liver. Particularly, EVs are essential carriers that contribute to the exchange of cellular information and transmit diverse pathophysiological statuses of the liver and other organs via systemic circulation (Fig. 1). In this review, we highlight recent observations on the role of EV-mediated intercellular communications in liver physiology and pathology with respect to both intrahepatic and interorgan interactions and discuss their potential therapeutic applications in liver diseases.
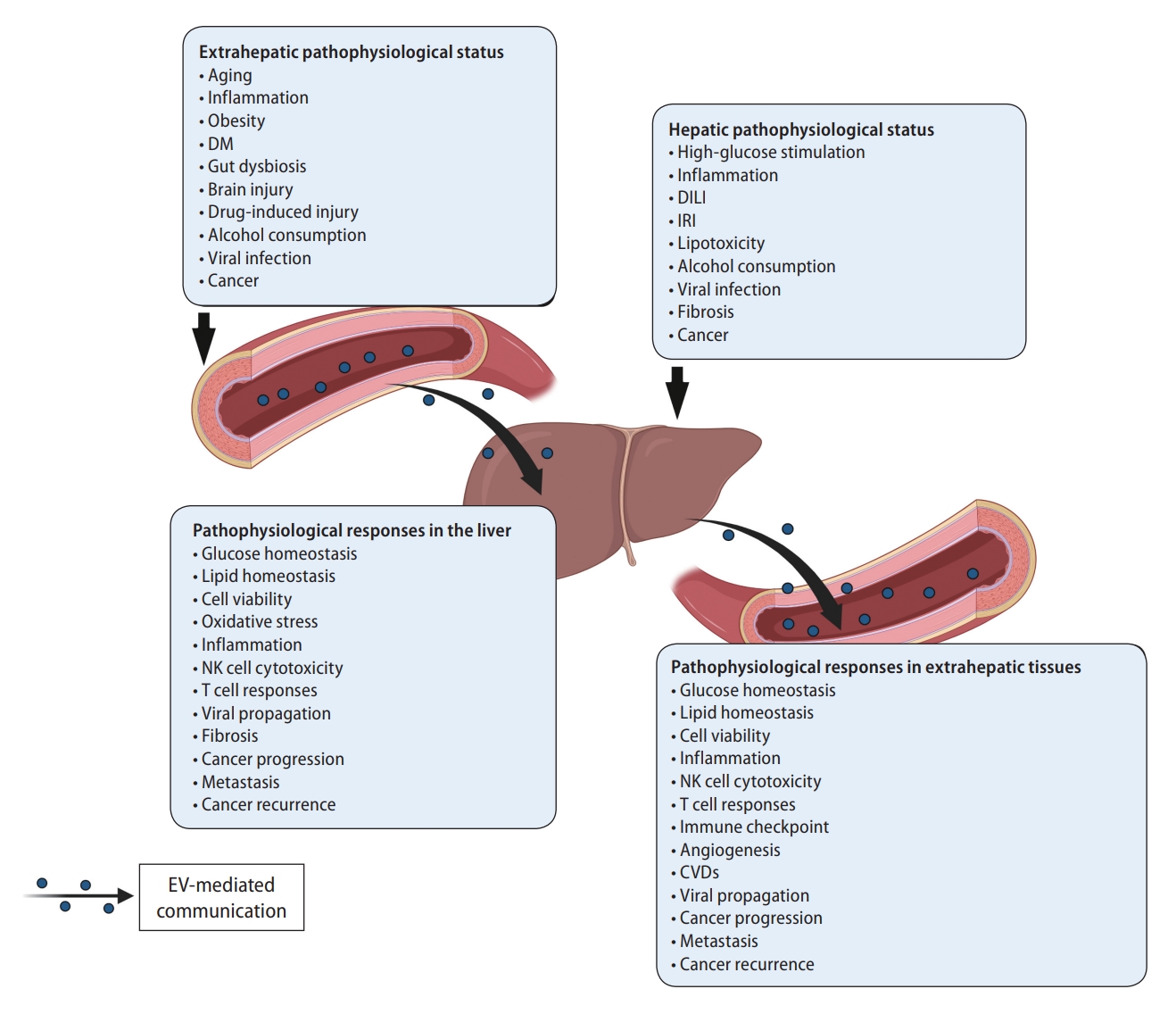
Circulating EVs affect liver physiology and pathology. Pathophysiological conditions in both the liver and extrahepatic tissues provoke increases in circulating EVs that often accumulate in the liver and modulate pathophysiological responses. Liver-derived circulating EVs target circulating immune cells and extrahepatic tissues to transmit their pathophysiological conditions. Systemic pathophysiological conditions including aging, inflammation, metabolic dysfunction, gut dysbiosis, injury, viral infection, and cancer aggravate liver physiology and promote pathological status through EV-mediated modulation of energy homeostasis, immune responses, disease-associated phenotypes, and cancer progression. In the same way, pathophysiological liver conditions including abnormal energy homeostasis, inflammation, injury, viral infection, fibrosis, and cancer promote pathological responses in the blood and distant organs by delivering their EVs through the circulation. Schematic illustration was created with BioRender.com. DM, diabetes mellitus; DILI, drug-induced liver injury; IRI, ischemia-reperfusion injury; NK, natural killer; CVD, cardiovascular disease; EV, extracellular vesicle.
EVs IN LIVER PHYSIOLOGY
EVs released from hepatic tissues
Numerous pathophysiological processes of the liver can be regulated by various free soluble factors, including fibroblast growth factors, platelet-derived growth factors (PDGFs), transforming growth factors, interleukins, and chemokines, produced by hepatocytes and hepatic non-parenchymal cells, including liver sinusoidal endothelial cells (LSECs), Kupffer cells, cholangiocytes (biliary epithelial cells), and hepatic stellate cells (HSCs) [12-14]. Similarly, EVs can package a diverse array of proteins, nucleic acids, and lipids in small membranous sacs and regulate various hepatic functions by delivering their cargo to recipient cells in a autocrine or paracrine manner. Most major cell types in the liver can exploit EVs for intrahepatic communication [15-17]. EVs contain numerous types of cargoes reflecting their cellular origin. Hepatocyte-derived EVs carry albumin and different enzymes, metabolizing nutrients, hormones, and xenobiotics [18]. Hepatocytes also release EVs harboring different types of RNAs that can regulate functions in other hepatic cells including HSCs [15], while EVs released from HSCs are captured by neighboring Kupffer cells and LSECs, inducing glycolysis (“Warburg effect”) [19] (Fig. 2A).

Liver-derived EVs mediate intercellular communication for regulating glucose homeostasis. EV-mediated intrahepatic and liver-involved inter-organ communications regulate metabolic homeostasis. Their EVs carry cargoes directly related to insulin sensitivity and energy metabolism. Depending on pathophysiological circumstances, they induce metabolic dysregulation, insulin resistance, and ultimately metabolic syndrome. (A) Activated HSCs deliver GLUT1 and PKM2 to KCs, LSECs and quiescent HSCs by utilizing EVs and stimulate glycolysis. (B) High-glucose stimulation enriches calpain 2 in EVs released from hepatocytes. These EVs deteriorate insulin sensitivity by cleaving insulin receptors. (C) EVs derived from miR-130a-3p-overexpressed hepatocytes alleviate energy homeostasis and insulin sensitivity in adipocytes by downregulating expression of PPARG and PHLPP2 genes. Schematic illustration was created with BioRender.com. LSECs, liver sinusoidal endothelial cells; KCs, Kupffer cells; GLUT1, glucose transporter 1; PKM2, pyruvate kinase M2; HSC, hepatic stellate cell; PHLPP2, PH domain and leucine-rich repeat protein phosphatase 2; PPARG, peroxisome proliferator-activated receptor gamma; EV, extracellular vesicle.
EV cargoes can be modified, along with changes in biological status of its sources, reflecting pathophysiological events in the liver. For example, profiles of cytochrome P450 (CYP) enzymes in hepatocyte-derived EVs change after drug-induced liver injuries [20]. In glucose homeostasis, calpain 2 loaded in EVs cleaves the ectodomain of the insulin receptor β subunit, which impairs insulin signaling in hepatocytes in an autocrine manner [21]. Notably, high-glucose stimulation can enrich calpain 2 in hepatocyte-derived EVs (Fig. 2B). In addition, disruption of the insulin receptor or downstream signaling can lead to insulin resistance and diabetic conditions [22]. Thus, glucose stimulation triggers the release of EVs from hepatocytes, which affects metabolic disease pathogenesis beyond their roles in regulating glucose homeostasis [21,22]. As described above, hepatocyte-derived EVs also carry transcripts or proteins related to drug-metabolizing enzymes [15,18,20]. After transfer to recipient cells, CYP enzymes and glutathione S-transferase seem to participate in the metabolism of drugs, reactive oxygen species (ROS), and lipids, as active enzymes themselves [23,24], although further studies need to be done.
EVs produced in hepatic cells are also delivered to various types of cells in distant tissues through the circulation [25,26]. The number of circulating ASPGR1-containing EVs, which are assumed to be released from hepatocytes, is positively correlated with impairments in glucose and lipid metabolism [27]. On the other hand, EVs derived from miR-130a-3p-overexpressing hepatocytes decrease fat accumulation and increase glucose uptake in adipose tissues, relieving insulin resistance in high-fat diet-fed mice by suppressing PHLPP2 (PH domain and leucine-rich repeat protein phosphatase 2) and PPARG (peroxisome proliferator-activated receptor γ) expression (Fig. 2C) [28]. Liver-derived EVs have thus both positive and negative implications for glucose homeostasis (Fig. 2). The liver also utilizes EVs to recruit immune cells from the circulation. After hepatic ischemia-reperfusion injury, hepatocytes release EVs containing oxidized phospholipids through the IRF1-Rab27a-mediated secretory pathway and recruit neutrophils in a toll-like receptor (TLR)4-dependent manner [29]. In addition, cholangiocytes release EVs encapsulating long non-coding RNA (lncRNA) H19 for recruiting bone marrow (BM)-derived macrophages by upregulating CCL2 and CCR2 expression in mouse cholestatic liver injury models [30]. Overall, diverse cell types in the liver activate and recruit circulating immune cells into the liver by delivering pro-inflammatory cargoes enclosed in EVs.
EVs delivered to the liver from extrahepatic tissues
EVs secreted from different tissues outside the liver also affect hepatic functions. EVs are present in almost all body fluids and are transported through systemic circulation, participating in various types of interorgan crosstalk [31-33]. In vivo imaging recently showed that EVs from the yolk syncytial layer are released into circulation and taken up by cells in distant organs in zebrafish embryos [34]. Moreover, circulating EVs are markedly increased in many pathological conditions such as obesity, diabetes mellitus, and cardiovascular diseases [35,36], and accumulate in the liver [8,37-40]. Hepatocytes and HSCs in chronically injured mouse liver tissues endocytose intravenously injected EVs obtained from serum of healthy mice [41]. In addition, Kupffer cells internalize circulating hemoglobin-containing EVs released from red blood cells [42]. Therefore, circulating EVs are considered important mediators that regulate diverse pathophysiological communications between the liver and other organs (Fig. 1), and may also serve as diagnostic biomarkers for different liver diseases.
Indeed, EVs derived from a variety of hepatic and extrahepatic tissues under different pathological conditions may reflect and modulate metabolic dysregulation in the liver. In the context of glucose homeostasis, plasma EVs derived from obese women downregulate glycogenesis and upregulate gluconeogenesis in hepatocytes cultured in vitro, even after stimulated by insulin [43]. In contrast to young BM-mesenchymal stem cell (MSC)-derived EVs, EVs released from aged BM-MSCs impair glucose homeostasis and promote insulin resistance by miR-29b-3p in metabolic organs, including the liver (Fig. 3A) [44]. On the other hand, microRNAs (miRNAs) in EVs derived from adipose tissue macrophages of lean mice can be taken up by hepatocytes and restore glucose homeostasis and insulin sensitivity in high-fat-diet-fed mice (Fig. 3B) [45]. In addition, gut microbiota-derived EVs can accumulate in the liver and promote insulin resistance in obese mice (Fig. 3C) [46].

EVs derived from extrahepatic tissues regulate hepatic glucose homeostasis. Extrahepatic tissues can deliver their pathophysiological information to the liver in the form of EVs through systemic circulation. Pathophysiological statuses related to metabolic dysfunction, such as aging and obesity increase the amount of metabolic homeostasis-disrupting molecules in these EVs and transmit metabolic dysfunctions of the donor to the liver by targeting essential factors for regulating glucose homeostasis and insulin sensitivity. (A) Aged BM-MSCs target SIRT1 gene expression in hepatocytes by transferring miR-29b-3p loaded in EVs and aggravate insulin resistance. (B) Opposed to lean ATM, obese ATM send miR-155-containing EVs to hepatocytes through circulation and repress PPARG gene expression, which is related to insulin sensitivity. (C) Gut microbial EVs are eliminated from blood by phagocytic activity of CRIg+ KCs in the liver. However, obesity reduces population of CRIg+ KCs and promotes the transport of microbial EVs containing pathogenic DNA to the liver by disrupting intestinal barrier integrity. It leads to aggravation of inflammation and insulin resistance in hepatocytes by activating cGAS/STING pathway. Schematic illustration was created with BioRender.com. BM-MSC, bone marrow mesenchymal stem cell; ATM, adipose tissue macrophages; PPARG, peroxisome proliferator-activated receptor gamma; CRIg, complement receptor of the immunoglobulin superfamily; KC, Kupffer cell; mDNA, gut microbial DNA; cGAS, cyclic GMP-AMP synthase; STING, stimulator of interferon genes; EV, extracellular vesicle.
In a similar way, immune responses in the liver are influenced by extrahepatic EVs. Focal inflammatory brain injury increases EV release from brain vascular endothelial cells, and these EVs can be taken up by the liver of normal recipient mice after injection, eliciting a systemic acute-phase response [40]. Liver natural killer T (NKT) cells are also one of the targets of EVs from distant organs. Intestinal mucosal cells-derived EVs deliver prostaglandin E2 to hepatic NKT cells and exert immunosuppressive effects [47]. It appears that extrahepatic EVs can modulate immune responses in pro- or anti-inflammatory directions depending on their cellular origin.
Extrahepatic EVs can cause or relieve toxicity in the liver according to the pathophysiological conditions of their sources. It was reported that plasma EVs contain more CYP2E1 than hepatocytes [48]. Accumulating evidence indicates that the levels of plasma CYP2E1-containing EVs are increased upon exposure to alcohol or acetaminophen and deteriorate hepatotoxicity [23,49,50]. In addition to CYPs, skin fibroblasts obtained from young human donors release EVs containing a high level of glutathione S-transferase Mu 2. These EVs also relieve oxidative stress in senescence-induced cells by modulating glutathione metabolism and ultimately rejuvenate various organs, including the liver, in aged mice [24].
EVs IN LIVER DISEASES
EVs derived from both hepatic and extrahepatic tissues affect progression (Table 1) or regression (Table 2) of liver diseases. Here we focus on the role of EVs in nonalcoholic steatohepatitis (NASH), alcoholic liver disease (ALD), viral hepatitis (VH), fibrosis, and HCC and highlight the potential therapeutic application of EVs against these liver diseases.
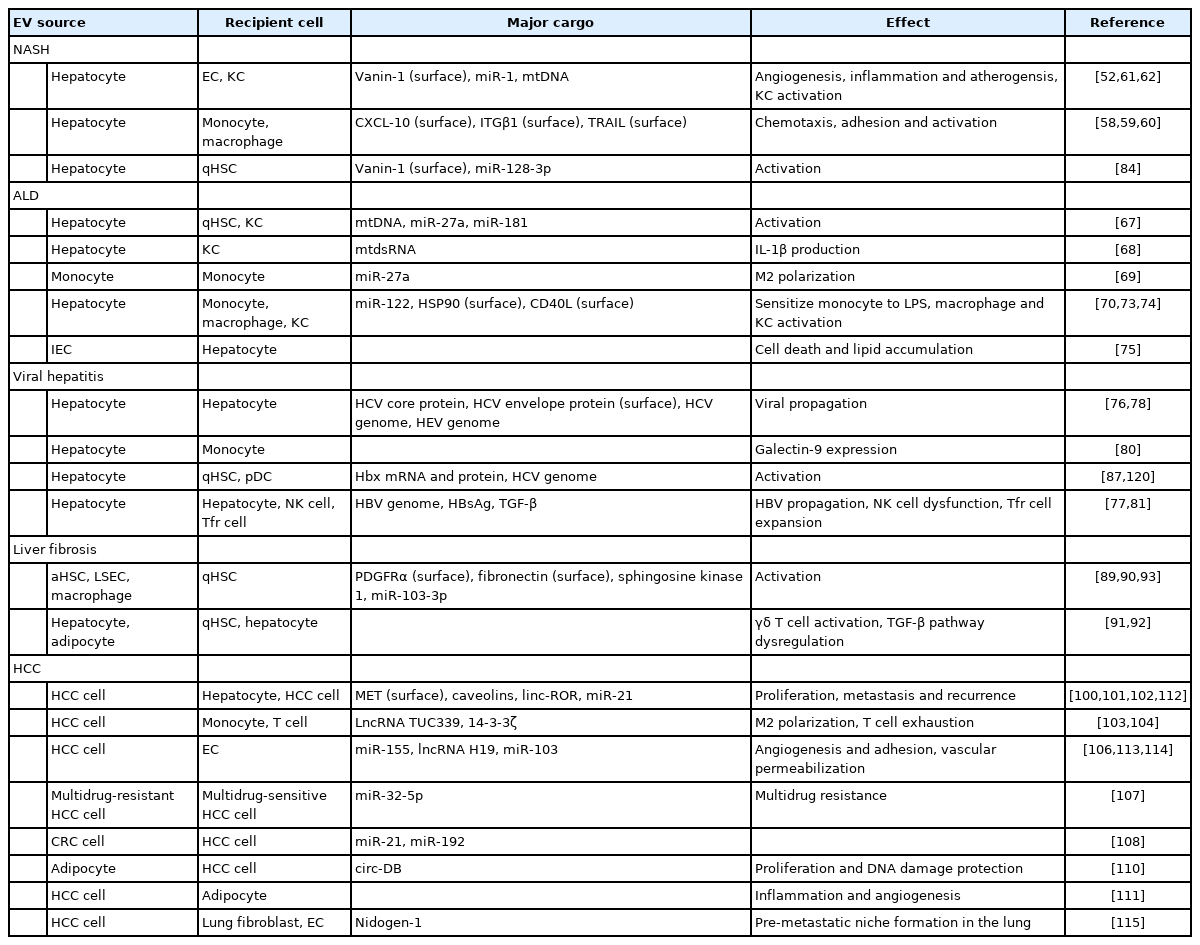
EV-mediated communication in progression of chronic liver diseases
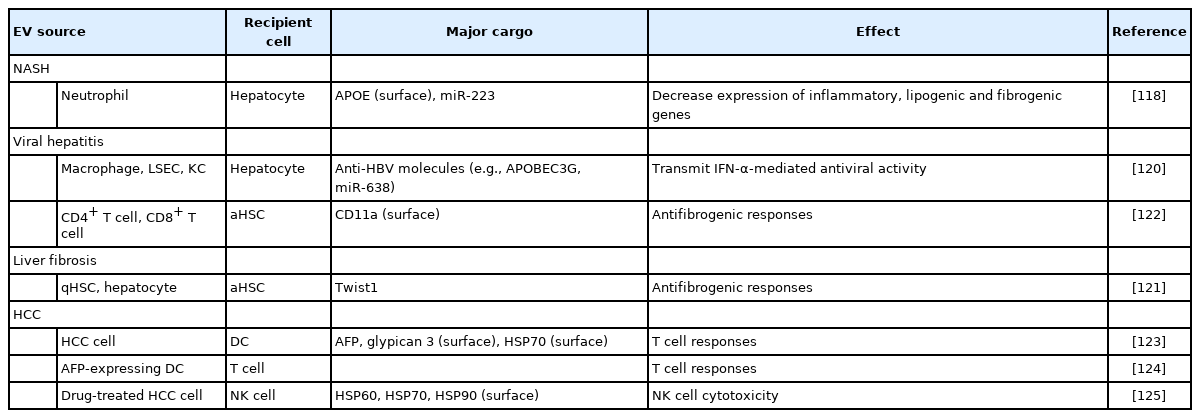
EV-mediated communication in regression of chronic liver diseases
EVs in NASH
Excessive free fatty acids induce cellular stress and dysfunctions in hepatocytes, causing hepatic steatosis and nonalcoholic fatty liver disease (NAFLD), which are major threats to health worldwide [51]. NASH is the advanced stage of nonalcoholic steatosis and is characterized by lipid accumulation with hepatic lobular inflammation, parenchymal cell death, and different degrees of fibrosis. These pathogenic events are mediated by intercellular interactions in various cell types in the liver and crosstalk between the liver and other organs [51].
EVs participate in a wide range of cellular interactions in NASH pathogenesis and progression. Hepatocytes release more EVs under lipotoxic conditions [52]. In line with this finding, EV levels are significantly increased in both the liver and blood of animals with NAFLD compared with normal animals, and the cargo profile changes with NAFLD progression [53]. EVs from subjects with NAFLD carry diverse proteins associated with inflammation, cell stress, and cell death, which are all representative features of NASH. Particularly, miR-122 and miR-192 levels in circulating EVs are highly elevated in mice with NAFLD, and these events are accompanied by significant reductions of these miRNAs in the liver [53]. Because both miRNAs are crucial regulators for lipid homeostasis, especially for inhibiting lipid accumulation, their decrease in the liver aggravates lipotoxicity and NASH [53-56].
Different types of immune cells play key roles in NASH pathogenesis. EVs released from hepatocytes under lipotoxic stress participate in intercellular communication between hepatic cells and immune cells. They recruit circulating immune cells into the liver tissue by delivering chemokines into the blood. For example, CXCL10, which chemoattracts macrophages, dendritic cells (DCs), NK cells, and T cells [57], are enriched in plasma EVs from murine NASH models [58]. Particularly, lipotoxic hepatocytes treated with palmitate or lysophosphatidylcholine increase the level of CXCL10-containing EVs in plasma, which then leads to mobilization of BM-derived macrophages into hepatic tissues [58]. In an animal model of NASH, integrin β1 is released from hepatocytes as a surface cargo of EVs and mediates adhesion between monocytes and LSECs, facilitating monocyte infiltration across the sinusoid to promote liver inflammation [59].
Circulating EVs secreted from lipotoxic hepatocytes can directly promote hepatic inflammation. Lipid-stimulated hepatocytes secrete tumor necrosis factor-related apoptosis-inducing ligand-containing EVs in a ROCK1-dependent manner (Fig. 4A, upper panel) and induce pro-inflammatory responses in BM-derived macrophages in vitro and mice with NASH [60]. In obese individuals with increased serum alanine aminotransferase levels, circulating EVs derived from hepatocytes contain elevated levels of oxidized mitochondrial DNA (mtDNA) and can activate TLR9, which is essential in the development of high-fat diet-induced NASH in mice [61]. In addition to promoting immunogenic environments inside the liver, EVs secreted from hepatocytes in a lipotoxic setting can be delivered through the blood circulation and foster inflammatory conditions in extrahepatic tissues. For example, EVs secreted from hepatocytes under NAFLD conditions induce miR-1-induced pro-inflammatory responses in endothelial cells by both nuclear factor-κB activation and Klf4 suppression, promoting atherosclerosis [62]. These data strongly suggest a potential link between NASH and cardiovascular disease. On the other hand, extrahepatic cell-derived EVs seem to participate in NASH progression. Serum EVs derived from diverse immune cells including macrophages, neutrophils, platelets, and invariant NKT cells show positive correlations with the severity of hepatic damage and NASH [63,64].
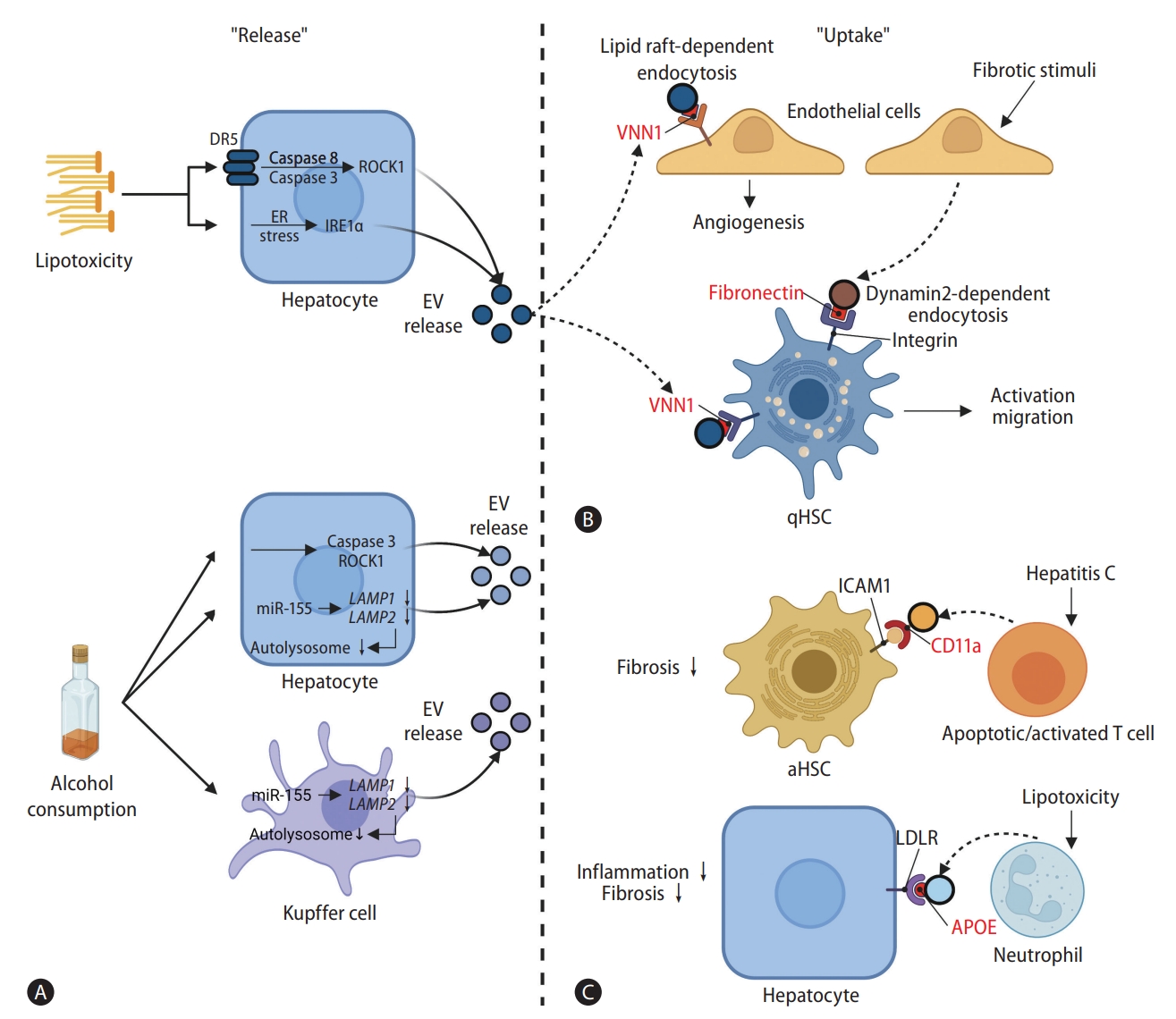
Release and uptake of EVs in pathological communications of hepatic and non-hepatic cells. EV exchanges between hepatocytes, HSCs, and immune cells are reinforced under pathological conditions induced by lipotoxic stresses and alcohol consumption. EV surface cargoes are highlighted in red color. (A) Lipotoxic stresses activate caspase 8/3 in hepatocytes by activating DR5 signaling in a ligand-independent manner, which leads to ROCK1 activation. Lipotoxicity also increases IRE1A activity by inducing ER stress. In consequence, ROCK1 and IRE1A both increase EV release from hepatocytes. Alcohol also induces caspase 3-mediated activation of ROCK1 in hepatocytes and decreases autophagic activities in hepatocytes and Kupffer cells via miR-155-mediated down-regulation of LAMP1 and LAMP2. These processes result in an increase in EV release from both hepatocytes and Kupffer cells. (B) EVs released from lipid-stimulated hepatocytes harbor VNN1 on their surface. VNN1 mediates EV uptake into LSECs through a lipid raft-dependent endocytosis, which aggravates fibrosis by promoting angiogenesis, and directly induces activation of HSCs. EV release from LSECs is promoted by fibrotic stimulation. The EVs are taken up by qHSCs via dynamin2-dependent endocytosis and worsen fibrotic conditions through HSC activation. The internalization of EVs by HSCs is mediated via fibronectin-integrin interaction. (C) Apoptotic or activated T cells in hepatitis C patients release EVs with CD11a on their surface. CD11a on the EV surface interacts with ICAM1 on aHSCs, facilitating EV internalization, which in turn alleviates fibrotic conditions. In lipotoxic conditions, EVs produced by neutrophils carry APOE on their surface. These EVs can be taken up by hepatocytes through LDLR and contribute to regression of fibrosis. Schematic illustration was created with BioRender.com. DR5, death receptor 5; ROCK1, rho-associated, coiled-coil-containing protein kinase 1; ER, endoplasmic reticulum; IRE1A, endoplasmic reticulum to nucleus signaling 1; EV, extracellular vesicle; LAMP, lysosomal-associated membrane protein; VNN1, vanin-1; qHSC, quiescent hepatic stellate cell; CD11a, integrin alpha L; aHSC, activated hepatic stellate cell; LDLR, low-density lipoprotein receptor; APOE, apolipoprotein E; LSEC, liver sinusoidal endothelial cell.
NASH may develop into liver cirrhosis and HCC, which both carry poor prognoses [65]. Further studies on the mechanisms underlying EV-mediated intercellular communication in NAFLD and NASH are needed to identify molecular targets for the development of therapeutic drugs, which are not yet available.
EVs in ALD
EV-mediated exchange of cellular information is involved in ALD development and progression. Alcohol targets a series of steps in the autophagy pathway and stimulates EV secretion from hepatocytes and Kupffer cells by suppressing autophagic functions through miR-155-mediated down-regulation of LAMP1 and LAMP2 [66] (Fig. 4A, lower panel). Specifically, EVs contribute to ALD pathogenesis and progression by modifying their cargoes with DNAs, miRNAs, and double-stranded RNAs. For example, hepatocytes isolated from a murine alcoholic hepatitis model release mtDNA-enriched EVs that are endocytosed and activate multiple pro-inflammatory cytokine genes in Kupffer cells through TLR9 activation [67]. In addition, mtdsRNA-enriched exosomes from alcohol-exposed hepatocytes stimulate TLR3-mediated interleukin (IL)-1β expression in Kupffer cells and trigger IL-17A production from γδ T cells at the early stage of ALD [68]. Alcohol-stimulated monocytes also utilize EVs containing M2-polarizing miR-27a to communicate with other monocytes [69]. EVs secreted from alcohol-treated hepatocyte deliver miR-122, sensitizing monocytes to lipopolysaccharides [70].
As expected, circulating EV levels are also increased in the plasma of both murine alcoholic hepatitis model and patients with ALD [67,69]. Similar cargo profiles, including miR-27a, miR-122, miR-155, and mtDNA, are observed in both EVs from the blood circulation and alcohol-exposed hepatocytes or Kupffer cells, indicating that the increased circulating EVs in ALD originate from the liver [69,71,72]. Circulating EVs in ALD are utilized to recruit and activate immune cells. In a murine ALD model, hepatocytes released heat shock protein (HSP) 90-containing EVs into the blood circulation, which promoted infiltration of monocytes and neutrophils into the liver [73]. ALD-associated EVs promote secretion of pro-inflammatory cytokines by directing macrophages toward the M1 inflammatory phenotype using their surface cargo, CD40 ligand [74]. Circulating EVs derived from tissues outside the liver also affect ALD progression. For example, EVs secreted from intestinal epithelial cells exert deleterious effects on hepatocyte survivability and lipid accumulation during acute alcohol injury [75]. In summary, EVs derived from hepatocytes, intrahepatic immune cells, circulating immune cells, and extrahepatic tissues all affect ALD pathology alone or in concert. The exact role of EVs in ALD is not fully understood, and many questions remained unanswered. However, the existing evidence provides novel insights into therapeutic approaches for ALD that attenuate the detrimental effects of ALD-associated EVs.
EVs in viral hepatitis
EVs derived from viral infected-hepatocytes can transmit the viral genome to neighboring hepatocytes. For example, the hepatitis C virus (HCV) core proteins and HCV genome have been detected in uninfected hepatocytes after treatment with EVs from HCV-infected cells, and the recipient hepatocytes can develop secondary infection [76]. Hepatitis viral genome-carrying EVs are also released into the circulation. Serum EVs harvested from patients with chronic hepatitis B infection harbor the hepatitis B virus (HBV) genome and can deliver it to uninfected hepatocytes [77]. EVs protect the viral genome from immune responses during delivery, so they can transmit the cargoes to recipient cells [78]. Inhibiting EV release from infected cells may therefore reduce hepatitis viral propagation [79].
EVs released from infected cells are then sent to immune cells associated with a viral infection. For instance, EVs from HCV-infected hepatocytes are delivered to monocytes and macrophages and regulate the expression of galectin-9 that induces HCV-targeting T cell apoptosis and regulatory T cell activation [80]. These EVs also increase the number of T follicular regulatory cells to suppress T follicular helper cells, which participate in humoral immunity [80,81]. HBV-infected cells exploit EVs to impair interferon (IFN)-γ production by NK cells and upregulate programmed death ligand 1 in peripheral blood monocytes [77,82]. In conclusion, EVs play critical roles in VH progression by mediating intercellular interactions between hepatocytes and immune cells.
EVs in liver fibrosis
Liver fibrosis is caused by excessive accumulation of extracellular matrix proteins and may progress to hepatic cirrhosis or liver cancer. Different CLDs including NASH, ALD, and VH may lead to liver fibrosis, and various paracrine factors secreted from multiple cell types are involved in progression and regression of liver fibrosis [83,84]. Activated HSCs are major sources of extracellular matrix production and scar formation in liver fibrosis. Recent findings provide useful insights into the interactions among key cells responsible for HSC activation and the development of liver fibrosis. However, the link between multicellular interactions and modulation of HSC phenotypes in liver fibrosis remains incompletely understood.
Hepatocytes release EVs harboring vanin-1 on their surface under lipotoxic conditions [85]. These EVs are enriched with miR-128-3p and activate HSCs after endocytosis by suppressing the expression of PPAR-γ, a key molecule for maintaining quiescent HSC phenotypes in the normal liver. Vanin-1-mediated uptake of lipotoxic EVs also stimulates hepatic angiogenesis (Fig. 4B), which is closely associated with fibrosis progression in CLDs [52,86]. ALD and VH can also progress to fibrosis by activating HSCs with EVs. Increased EV secretion from hepatocytes in mice with alcoholic hepatitis activates HSCs, in part by miRNA-mediated repression of quiescent phenotypes [67]. HBV-infected hepatoma cells release EVs containing HBx mRNA and protein, which induce the expression of profibrotic markers and proliferation signaling in quiescent HSCs [87].
EVs from activated HSCs cause dysfunction in different types of liver cells including quiescent HSCs, LSECs, and Kupffer cells [19]. PDGF-BB-induced activation of HSCs promotes in vitro migration of quiescent HSCs and in vivo liver fibrosis through delivering PDGFRα-enriched EVs via the SHP2-mediated pathway [88,89]. EVs derived from PDGF-BB-treated HSCs also carry hedgehog, which activates LSECs in the setting of fibrosis [16]. Reciprocally, LSECs can deliver EVs to induce HSC activation. Following fibrotic stimuli, LSECs upregulate sphingosine kinase 1 mRNA expression, resulting in enhanced production of sphingosine 1-phosphate and HSC chemotaxis [90]. Additionally, sphingosine kinase 1-enriched EVs secreted from LSECs are delivered to HSCs and accelerate their activation by activating Akt signaling and producing sphingosine 1-phosphate [90]. In animal models of CLD, hepatocyte-derived EVs promote HSC secretion of IL-17, IL-1β, and IL-23 in a TLR3-dependent manner and induce differentiation of naïve T cells into γδ T cells, thereby aggravating liver fibrosis [91].
Circulating EVs derived from extrahepatic cells are also important regulators of liver fibrosis development and progression. For example, EVs from human obese visceral adipose tissues dysregulate transforming growth factor-β signaling toward pro-fibrogenic phenotypes in hepatocytes and HSCs [92]. EVs released from lipopolysaccharide-stimulated THP-1 macrophages carry miR-103-3p, which is abundant in serum EVs from patients with liver fibrosis, and activate HSCs in vitro by targeting KLF4 mRNA [93]. In the context of NASH or ALD, circulating EVs in liver fibrosis show similar patterns of cargo enrichment and activities with activated HSC-derived EVs [16,89]. In addition, fibrosis-associated EVs from HSCs can be delivered to immune cells and modulate a series of inflammatory responses [94]. Thus, it can be speculated that activated HSCs also release EVs into the circulation and modulate pathological conditions including inflammation in other organs outside the liver. As described above, EVs from different hepatic cells and extrahepatic tissues contribute to liver fibrosis progression. Therefore, understanding the complex intercellular crosstalk is essential to design effective therapeutic approaches for liver fibrosis.
EVs in HCC
HCC is a leading cause of cancer-associated deaths worldwide and has a poor survival rate. EVs seem to actively participate in pathologic communications during the progression of CLDs to HCC. In aldehyde dehydrogenase 2-deficient mice, chronic alcohol exposure elicits oxidized mtDNA-enriched EV release from hepatocytes [95]. These EVs can be internalized by HCC cells and induce both ROS production and oncogenic signal transduction. HCV infection increases the level of serum-circulating EVs harboring differentiation antagonizing non-coding RNA, which is positively correlated with HCC recurrence [96]. Specifically, miR-155, which is an emerging factor contributing to CLD progression to HCC, is significantly enriched in serum EVs from mice and patients with CLD [71,97-99].
In HCC development and progression, EVs have crucial roles as intratumoral messengers. Firstly, HCC exploits EVs to transform hepatocytes. HCC cells increase the proliferation, survival, and migration of normal hepatocytes by delivering their EVs containing protumorigenic RNAs and proteins, as well as lncRNA regulators of reprogramming [100,101]. HCC cells also utilize EVs to interact with each other in the HCC population. Tumor-derived EVs stimulate proliferation of HCC cells, thereby enhancing tumor formation by targeting PTEN with miR-21 [102]. Interaction between tumor cells and tumor microenvironment through EVs exacerbates HCC progression. HCC-derived EVs polarize macrophages to M2 phenotype, similar with tumor-associated macrophages, partly by delivering lncRNA TUC339 [103]. HCC cells also impair anti-tumor activities of CD8+ T cells through releasing EVs. HCC cells can directly deliver 14-3-3ζ to CD8+ T cells in an EVs-encapsulated form, which induces expression of inhibitory receptors such as programmed death-1 and TIM-3 [104].
Angiogenesis is a crucial process in HCC growth. HCC continuously induces abnormal angiogenesis, resulting in extensively leaky vasculature with abnormal blood flow that leads to hypovascularized areas and severe hypoxia [105]. HCC cells directly deliver miR-155-containing EVs to endothelial cells to stimulate angiogenesis under hypoxic conditions [106]. Advanced-stage HCC patients are currently treated with vascular epithelial growth factor receptor-targeting drugs such as sorafenib and sunitinib to inhibit angiogenesis [105]. However, miR-32-5p can be delivered through EVs and induces multidrug resistance in HCC by promoting epithelial-mesenchymal transition (EMT) and angiogenesis [107].
In addition to the roles of EVs in intratumoral communication, EVs from extrahepatic tissues affect HCC development and progression. EVs can exchange cellular information between different types of cancer cells. EVs from colorectal cancer cells or HCC stem-like cells contain diverse RNAs, including miR-21 and miR-192, which are associated with HCC progression, and transfer their cargoes to HCC cells [102,108,109]. One group reported that EVs carrying circular RNAs related to deubiquitination (circ-DB) are released from adipocytes [110]. These circ-DB-enriched EVs are internalized by HCC cells and activate ubiquitin-specific protease (USP)7 by repressing miR-34a. USP7 is upstream of cyclin A2 and DNA repair. Therefore, EVs released from adipocytes promote HCC progression by supporting proliferation and reducing DNA damage [110]. Conversely, HCC cells send EVs to adipocytes and transform their phenotypes into pro-inflammatory, pro-angiogenic, and pro-tumorigenic statuses [111].
HCC metastasis is also mediated by EVs. Highly metastatic HCCs secrete EVs that induce EMT in low metastatic HCC cells by activating MAPK/ERK signaling [112]. Adhesion of cancer cells to endothelial cells is important for both intravasation and extravasation. CD90+ HCC cells convey lncRNA H19-containing EVs to endothelial cells, which stimulates adhesion between HCC and endothelial cells by enhancing adhesion molecule expression [113]. In addition, EVs enriched with miR-103 from HCC cells increase vascular permeability by weakening vascular junctions, leading to increased intravasation and extravasation of hepatoma cells [114]. Finally, circulating EVs derived from HCC build pre-metastatic niches, preferentially in the lung by activating angiogenesis and lung fibroblasts to express tumor necrosis factor receptor 1 to support HCC colonization, growth, migration, and invasion [115]. EVs also mediate HCC recurrence. MiR-92b is highly enriched in serum EVs of HCC patients and partly contributes to HCC recurrence after living donor liver transplantation by suppressing NK cell-mediated cytotoxicity [116]. Similarly, EVs derived from highly metastatic hepatoma cells promote HCC recurrence by facilitating EMT through the MAPK/ERK signaling pathway [112].
EVs are emerging as an important mediator of intercellular communications in HCC progression that deliver RNAs and proteins between HCC cells, immune cells, and other extrahepatic cells. Further studies investigating the biogenesis and trafficking of HCC-associated EVs and identifying their oncogenic cargoes are urgently needed to develop novel therapeutic methods for inhibiting HCC progression, metastasis, and recurrence.
EVs to promote liver disease regression
EVs have conflicting dual roles in liver pathology progression. As mentioned above, EVs derived from pathologic hepatic tissue aggravate liver diseases by interacting with numerous types of cells through diverse mechanisms. However, they can also suppress activities associated with liver disease progression. Indeed, EVs isolated from both normal and acute-injured liver tissues exert regenerative effects partly by inducing hepatocyte growth factor expression to reduce apoptosis and promote hepatocyte proliferation in a murine acute liver injury model [117].
EVs can drive the regression of CLDs. Under NASH conditions, liver-infiltrating neutrophils can downregulate inflammatory gene expressions in hepatocyte by transferring EVs containing miR-223 [118]. This event is mediated by interactions between the low-density lipoprotein receptor and apolipoprotein E on hepatocytes and EVs derived from neutrophils (Fig. 4C). EVs are also utilized for viral genome propagation. However, viral genome-containing EVs can be taken up by plasmacytoid DCs and induce innate immune responses including IFN-α secretion [119]. Subsequently, secreted IFN-α can stimulate hepatic non-parenchymal cells to release EVs that deliver anti-viral molecules to hepatocytes [120].
EVs are also able to prevent fibrosis development in CLDs. As described above, infiltrating neutrophils release miR-223-containing EVs, which can be taken up by hepatocytes and attenuate NASH progression to fibrosis by inhibiting the expression of fibrogenic genes [118]. In addition, EVs secreted from quiescent HSCs and hepatocytes contain high levels of Twist that promotes miR-214 expression and downregulates connective tissue growth factor, which is responsible for the production of fibrogenic molecules [121]. In HCV-associated liver fibrosis, T cell-derived EVs can be internalized into HSCs in an intercellular adhesion molecule-1-mediated manner and induce the expression of anti-fibrotic genes while repressing pro-fibrogenic genes [122] (Fig. 4C). Circulating EVs also participate in liver fibrosis regression by modulating hepatocyte proliferation and cell death, HSC activation, and inflammatory responses [41].
HCC progression is also affected by HCC-derived EVs that carry an array of tumor antigens. DCs treated with HCC-derived EVs induce a strong T cell-mediated immune response in vitro and in vivo, resulting in a significant retardation of tumor growth [123]. These immune responses, including regulatory T cell recruitment and anti-inflammatory cytokine secretion, are also mediated by EVs released from DCs [124]. Following treatment of HCC with anti-cancer drugs, hepatoma cells release EVs containing HSPs on their surface, such as HSP60, HSP70 and HSP90 [125]. These HSP-bearing EVs enhance NK cell cytotoxicity and granzyme B secretion, improving host immune responses against tumor cells.
Therapeutic potentials of EVs
As described above, EVs are crucial mediators for a wide range of intercellular communication underlying liver disease development, progression, and regression. Several enzymes, including nicotinamide phosphoribosyl transferase, are exclusively transferred by EVs from donor cells to recipients [126,127]. Therefore, manipulating EV-mediated crosstalk could be a promising therapeutic strategy to treat different types of liver diseases.
One of the approaches for modulating EV-mediated interactions is changing the cargo inside before it is released. In a murine model of NASH, CXCL10 becomes enriched in EVs through an MLK3-dependent pathway [58]. After deletion of MLK3, CXCL10 levels decrease in these EVs, which reduces hepatic inflammation and injury. Blocking the biogenesis and release of specific EVs are effective ways to disturb EV exchanges. Lipotoxicity-induced hepatic inflammation and injury are attenuated by inhibiting endoplasmic reticulum to nucleus signaling 1, which is known to stimulate EV release upon endoplasmic reticulum stress [128]. Disrupting caspase 3 or ROCK1 activities suppresses alcohol-induced EV secretion and reduces macrophage-mediated hepatic inflammation [74] (Fig. 4A). Once EVs are secreted from cells, hindering their adhesion to and internalization by recipient cells may impede disease progression. Pro-fibrotic activation of HSCs by EVs released from LSECs is hampered by disturbing fibronectin-integrin interactions between the surfaces of EVs and HSCs or inhibiting dynamin-mediated endocytosis [90] (Fig. 4B). Vanin-1 is an EV surface molecule that is an important target for suppressing EV uptake by HSCs to treat liver fibrosis [52,85].
EVs can be exploited to deliver therapeutic molecules to liver tissues. As mentioned above, systemically injected EVs preferentially accumulate in the liver [37-40]. Furthermore, when they are delivered through the circulation, the lipid bilayer of EVs protects cargo molecules against enzymes including proteases and nucleases [129]. Thus, EVs isolated from both hepatic and extrahepatic cells can be injected to treat CLDs. For instances, systemically injected EVs derived from hepatic progenitor cells attenuate inflammation and fibrotic phenotypes in the NASH liver [130]. MSC EVs have also been extensively investigated for their ability to regress liver disease [131]. In addition to EVs obtained from BM-MSCs to promote liver regeneration [131], tonsil MSCs-released EVs enriched with miR-486-5p also attenuate liver fibrosis by inhibiting hedgehog signaling after intravenous administration to CCl4-induced liver fibrosis mice [132].
EVs can be engineered to enhance their sustainability and increase the amount of therapeutic cargo molecules. Embedding MSC-derived EVs in polyethylene glycol hydrogels prolongs EV retention in the body [133]. Upon injection, EVs can be continuously and progressively leaked from swollen hydrogels for up to 30 days in peritoneum and mainly accumulate in the liver, whereas regular EVs without hydrogels disappear within 7 days after injection [133]. EVs have been utilized to deliver specific molecules of interest to the liver by overexpressing miRNAs or proteins in EV-producing cells. For example, insulin-like growth factor (IGF)-I can be increased in EVs from human umbilical cord perivascular cells by overexpression [134]. These engineered EVs transfer a high amount of IGF-I to the liver and significantly reduce liver fibrosis in mice. Engineering EVs to contain specific molecules might also be useful to develop innovative vaccination strategies. 293T cell-derived EVs engineered to upload the full-length HCV-N53 protein produce memory CD8+ T cells when injected in mice, suggesting that the engineered EVs can boost CD8+ T cell immunity in HCV-infected patients [135]. Different proteins responsible for disease progressions can be targeted by EVs engineered to carry specific small RNAs. EVs carrying small interfering RNA directly targeting β-catenin are used for inhibiting tumor growth and promoting therapeutic efficacy of anti-programmed death-1 antibody in HCC [136]. Activities of nucleic acids in recipient cells can also be regulated by EVs. B cell-derived EVs can function as vehicles to transfer miR-155 mimics or inhibitors to hepatocytes or macrophages and reduce their inflammatory responses [137]. In addition, CRISPR/dCas9-VP64 system loaded in EVs can be delivered into HSCs and activates hepatocyte nuclear factor 4 alpha gene expression, leading to reprogramming of HSCs toward hepatocytes [138]. Lastly, EVs can be loaded with synthetic or natural compounds and delivered to the liver. For example, the anti-cancer drug norcantharidin (NCTD) can be encapsulated in BM-MSC-derived EVs by electroporation [139]. These NCTD-carrying EVs have in situ homing effects on tumor sites and non-toxically reduced tumor growth in a mouse model of HCC.
CONCLUSION
EVs have emerged as crucial messengers of intercellular crosstalk and are attracting attention due to their multifaceted roles in diverse physiological and pathological processes of many organs, including the liver. Growing evidence has implicated EVs in communications between different hepatic cell types and between the liver and other organs under different pathological conditions. However, a broader understanding of how EVs contribute to pathological and therapeutic processes in the liver is essential to develop novel EV-based strategies for liver disease diagnosis and treatment. Although many previous studies have investigated the roles of EVs in various hepatic and extrahepatic cells, the molecular mechanisms underlying EV biogenesis, release, uptake, and intracellular processes remain poorly understood, partly due to technical hurdles associated with EV preparation and analysis. Given the important roles of EVs in the progression or regression of different liver diseases, the multiple steps required for EV-associated extra- and intercellular processes could be prospective molecular targets to achieve effective therapeutic outcomes. To this end, more precise investigations should be conducted to identify key therapeutic cargoes enclosed in EVs. Particularly, EVs derived from hepatic and extrahepatic cells contain various molecules reflecting their cellular origins, and the concentration and composition of EV cargo can be changed depending on pathophysiological conditions of EV-producing cells. Therefore, careful analyses of the cargo molecules inside and on the surface of EVs and identification of EV mechanisms of action under normal and pathologic conditions will facilitate therapeutic application of EVs in the clinical setting without raising safety issues. There is also a need for effective engineering strategies to improve EV production and targeting ability and to modify EV cargoes for efficient delivery of therapeutic molecules to the appropriate cells in injured liver tissue. Despite technical limitations and insufficient knowledge about EV cargoes, biogenesis, release, and uptake, they have great potential as both targets and tools in innovative cell-free EV-based therapies for currently incurable liver diseases.
Notes
Authors’ contributions
Y.L. contributed to the manuscript drafting and preparation of the figures. J.H.K. reviewed and wrote the manuscript.
Conflicts of Interest
The authors have no conflicts to disclose.
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) Grant funded by the Korean Ministry of Science and ICT (No. 2020R1A2C2006240).
Abbreviations
ALD
alcoholic liver disease
BM
bone marrow
CLD
chronic liver disease
CYP
cytochrome P450
DC
dendritic cell
EMT
epithelial-mesenchymal transition
EV
extracellular vesicle
HBV
hepatitis B virus
HCC
hepatocellular carcinoma
HCV
hepatitis C virus
HSC
hepatic stellate cell
HSP
heat shock protein
IFN
interferon
IGF
insulin-like growth factor
IL
interleukin
IRE1A
inhibiting endoplasmic reticulum to nucleus signaling 1
lncRNA
long non-coding RNA
LSEC
liver sinusoidal endothelial cell
miRNA
microRNA
MSC
mesenchymal stem cell
mtDNA
mitochondrial DNA
NAFLD
nonalcoholic fatty liver disease
NASH
nonalcoholic steatohepatitis
NCTD
norcantharidin
NKT
natural killer T
PDGF
platelet-derived growth factor
ROS
reactive oxygen species
TLR
toll-like receptor
USP
ubiquitin-specific protease
VH
viral hepatitis