Subclinical versus advanced forms of alcohol-related liver disease: Need for early detection
Article information

Abstract
Alcohol-related liver disease (ALD) consists of a wide spectrum of clinical manifestations and pathological features, ranging from asymptomatic patients to decompensated cirrhosis and hepatocellular carcinoma. Patients with heavy alcohol intake and advanced fibrosis often develop a subacute form of liver failure called alcohol-induced hepatitis (AH). Globally, most patients with ALD are identified at late stages of the disease, limiting therapeutic interventions. Thus, there is a need for early detection of ALD patients, which is lacking in most countries. The identification of alcohol misuse is hampered by the existence of alcohol underreporting by many patients. There are useful biomarkers that can detect recent alcohol use. Moreover, there are several non-invasive techniques to assess the presence of advanced fibrosis among patients with alcohol misuse, which could identify patients at high risk of liver related events or early death. In this review, we discuss differences between early stages of ALD and AH as the cornerstone of advanced forms. A global overview of epidemiological, anthropometric, clinical, analytical, histological, and molecular differences is summarized in this article. We propose that campaigns aimed at identifying patients with subclinical forms can prevent the development of life-threatening forms.
INTRODUCTION: PREVALENCE OF ALCOHOLRELATED LIVER DISEASE
Alcohol consumption is one of the most frequent causes of liver disease worldwide. In 2016, according to the World Health Organization, the harmful consumption of alcohol resulted in 3 million deaths (5.3% of all deaths) worldwide and 132.6 million disability-adjusted life years (DALYs) – i.e., 5.1% of all DALYs in that year. Alcohol-related mortality is more prevalent in men and higher than other important diseases such as tuberculosis, acquired immunodeficiency syndrome and diabetes [1].
Diagnosis of alcohol-related liver disease (ALD) requires documentation of an alcoholic use disorder (AUD) and exclusion of other causes of liver disease. ALD consists of a wide spectrum of clinical manifestations and pathological features, from asymptomatic patients to decompensated cirrhosis and hepatocellular carcinoma (Fig. 1) [2-5]. The early stage of the disease is not well understood, and there is a need to better understand its natural history, risk factors of progression, and noninvasive diagnosis biomarkers [3,5-7]. Therefore, underdiagnosis is the rule until severe forms like alcohol-induced hepatitis (AH) develop and mortality is high despite abstinence. In this stage, patients have a terrible prognosis, with short-term mortality rates as high as 50% at three months due to subsequent organ failure and acute-onchronic liver failure (ACLF) [7,8].
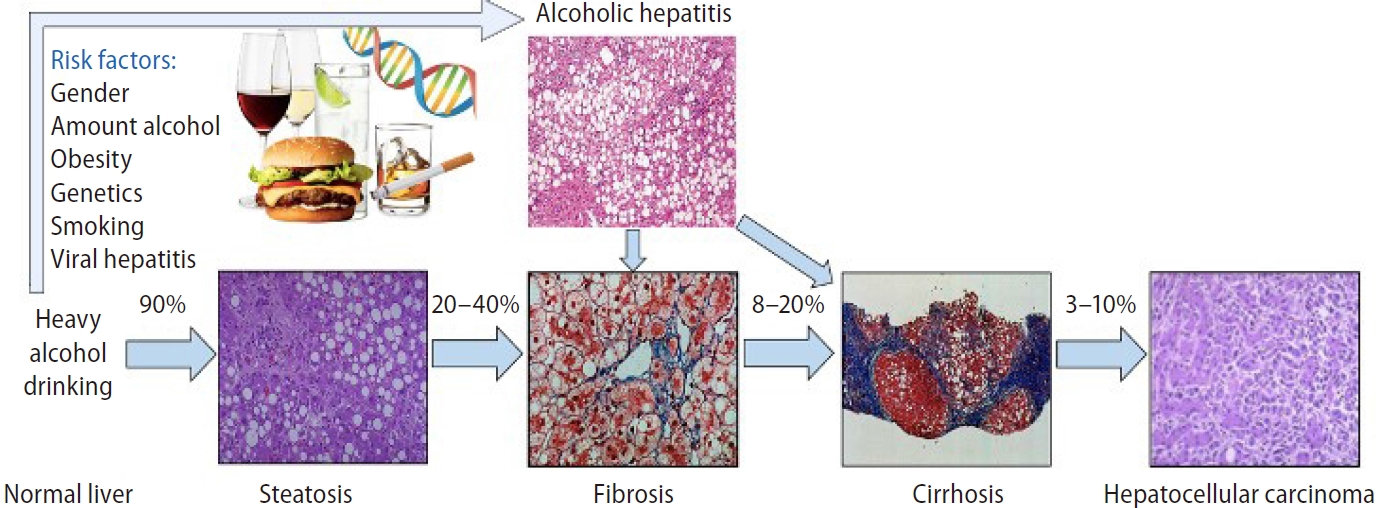
Spectrum of alcohol-related liver disease. The numbers represent the percentage of patients with progression. From Yamada’s Textbook of Gastroenterology. Permission for their use from the publisher.
The histological and clinical features observed in patients with severe ALD are difficult to replicate in animal models. Additionally, the difficulty in conducting clinical trials in patients with active AUD, the social stigmatization and marginalization of this population, the lack of interest from drug companies and the limitations of current experimental models contribute to a slow progress in ALD management. Thus, the treatment of patients with ALD hasn’t changed much in recent decades, and there aren’t any targeted and personalized medicines available [2]. To date, the most effective therapy to attenuate the clinical course of ALD and even reverse histological injuries is prolonged alcohol abstinence [4].
Identifying risk factors in individuals with AUD that predispose to ALD development is crucial for implementation of public health policies and reduce morbimortality associated to ALD. Several significant biological factors and possible therapeutic targets have been explored in recent translational research. The early recognition of early stages of ALD with subsequent behavioral therapies ought to be encouraged in primary care settings [2,6,8-10]. For example, alcohol screening questionnaires and basic laboratory test including hepatic profile in high-risk patients.
In this review, we will differentiate between early forms of ALD and AH, the most severe form of ALD, from the epidemiological, anthropometric, clinical, analytical, histological, and molecular stand of points.
DISEASE STAGES AND NATURAL HISTORY
ALD includes a wide range spectrum of early and advanced phenotypes. Early phenotypes do not usually have symptoms and can be categorized as subclinical forms. Subclinical stages include fatty liver disease or steatosis, steatohepatitis (ASH) with or without fibrosis and compensated cirrhosis. Although non-invasive biomarkers can have a role in steatosis and fibrosis diagnosis, histological evaluation is needed to define these subclinical stages. Patients with subclinical liver disease and persistent active drinking can end up developing advanced forms of ALD. Advanced stages are symptomatic and entail a poor prognosis due to liver-related complications. This phenotype includes AH, ACLF and decompensated cirrhosis (Fig. 2).
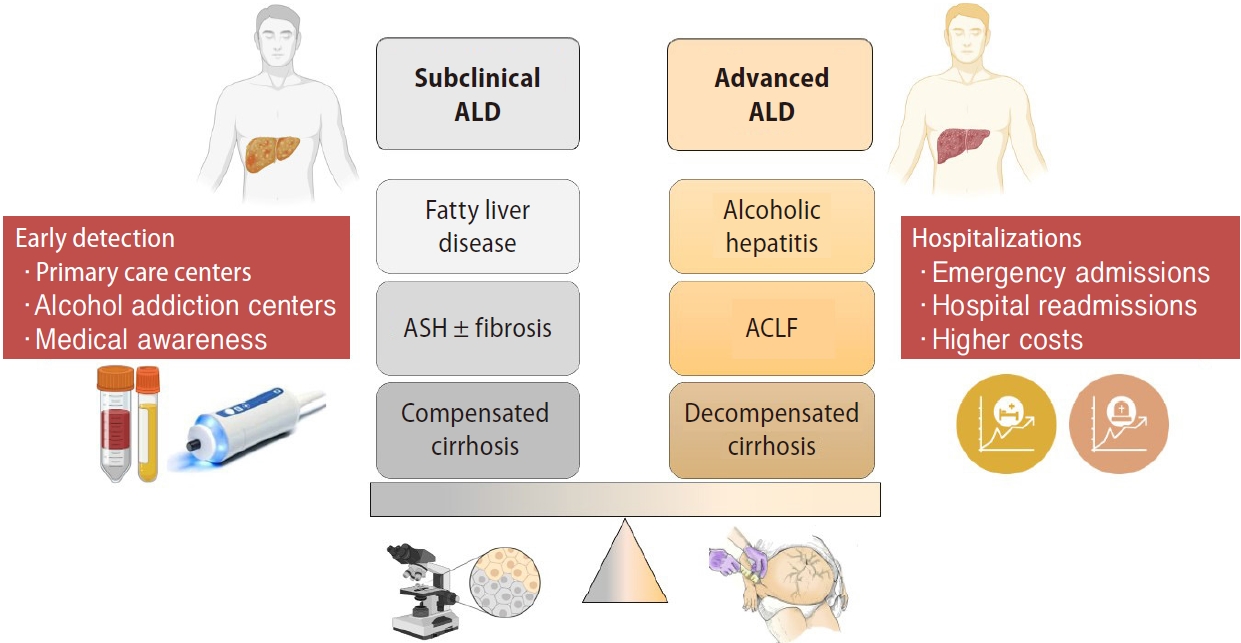
Subclinical versus advanced forms of ALD. ALD, alcohol-related liver disease; ASH, steatohepatitis; ACLF, acute-on-chronic liver failure.
Steatosis can develop as soon as 3 to 7 days after heavy alcohol consumption. Steatosis is mostly asymptomatic and may be associated with mild elevation of gamma-glutamyltransferase (GGT). It is histologically characterized by macrovesicular fat accumulation, typically located in centrilobular areas. Simple steatosis should not be considered a benign condition since it increases ALD annual mortality up to 6% [11]. In the same line, mortality in biopsy confirmed non-alcoholic fatty liver disease (NAFLD) is also increased among simple steatosis stages [12]. Although different medical conditions, both ALD and NAFLD show that steatosis carries prognostic implications by itself. Continuous and excessive alcohol drinking may lead, in 10–35%, to the development of ASH, which is characterized by steatosis, hepatocellular damage (i.e., ballooning, Mallory-Denk bodies [MDB]), inflammatory infiltrates–mainly neutrophils–, and different degrees of fibrosis with a pericellular pattern distribution [6]. Approximately 20–40% of patients with ASH will develop progressive fibrosis, of which 8–20% will develop cirrhosis. The risk of cirrhosis is increased in patients with ASH on biopsy as compared with patients with simple steatosis [13]. Once cirrhosis is established, its natural history is characterized by an asymptomatic compensated phase followed by a decompensated phase, marked by the development of overt clinical signs, the most frequent of which are ascites, bleeding, encephalopathy, and jaundice (Fig. 2) [14].
When persistent alcohol intake is maintained, an episode of AH can be developed. Till date, there is no clear explanation why some patients develop this phenotype, nor what are the triggers. It has been speculated that this is due to an increased alcohol consumption, but this has not been firmly demonstrated. AH is associated with high mortality, which can reach 50% in three months, and the median survival time of patients with advanced liver cirrhosis can be as low as 1–2 years [15].
In a recent study, it was demonstrated that the presence of advanced fibrosis and continued alcohol consumption were the main parameters associated with early mortality in patients with compensated forms of ALD [4]. Moreover, a new parameter has been introduced recently to quantify fibrosis in liver biopsies using digital image analysis, the collagen proportionate area [16]. This parameter predicts liver-related mortality in ALD and hepatic decompensation and/or liver-related death in early/compensated ALD [17].
SUBCLINICAL ASH VS. AH: CLINICAL, ANALYTICAL AND HISTOLOGICAL FEATURES
Most patients with ALD are identified during late stages of the disease when liver decompensation occurs and when mortality is high despite ethanol cessation. In fact, a global epidemiologic study in 2017 [18] showed that ALD is by far the liver disease etiology that is detected at the latest stages. These results strongly suggest that there is a dire need for the early detection of ALD patients, which currently is almost nonexistent. Recently, a study comparing ASH and AH disease stages has been conducted. This study concluded that patients with AH had higher liver failure and mortality compared to ASH patients (50% vs. 10% 1-year mortality, respectively) with significant different clinical, histological and molecular features [19].
Although it is assumed that ASH is the harbinger of transition to severe forms of ALD, there are few studies that have assessed clinical and histological features that predict liver disease progression in these patients [3,4,20-22]. Much more research attention has been paid to the severe form of AH [23-26]. Currently ASH can only be diagnosed with liver biopsy. Similar to NAFLD, there are no signs, symptoms, or biochemical tests that allow the confident diagnosis of ASH [14]. Although asymptomatic, identification of subclinical, molecular and histologic features of ASH would favor its detection worldwide.
The diagnosis of ALD is based on history of heavy alcohol use, typical laboratory markers and clinical features [5]. Compared to similar diseases such as NASH, few patients with ALD undergo a liver biopsy. Most patients with both early and advanced forms are diagnosis without histological assessment. The recent development of a specific fibrosis grading system for patients with ALD could stimulate this field [27]. In patients with AH, a transjugular liver biopsy is justified when there are confounding factors [28], and the histological changes (i.e., presence advanced fibrosis, polymorphonuclear infiltration, etc.) predict short-term survival [23].
Some methods to determine alcohol as the major etiology of liver disease include the medical history, surveys, physical examination and laboratory test. It is also important to consider second liver hits that can influence ALD prognosis.
History of alcohol misuse
Criteria from the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition [29], are used to define AUD and to determine its severity. Severity is based on the number of criteria a person meets based on their symptoms in the past year. Some surveys can help us to distinguish patients with alcohol abuse. The Alcohol Use Disorders Identification Test (AUDIT) comprises ten questions with a specific scoring system to diagnosis alcohol abuse (AUDIT score >8) and alcohol dependence (AUDIT score >20) [30] with 73% sensitivity and 91% specificity vs. 85% sensitivity and 89% specificity respectively [31]. The CAGE questionnaire is also a useful and an easily applied tool. It is more sensitive than the AUDIT to detect alcohol abuse and dependence, but is less effective in recognizing non-severe drinking disorders [32]. However, many patients tend to underreport, particularly in the pre- or post-liver transplant interval, for fear of reprisal by the transplant program. The Clinical Institute Withdrawal Assessment for Alcohol (CIWA) scale assesses the severity of alcohol withdrawal. A randomized, double blind trial published in JAMA in 1994 showed that management for alcohol withdrawal that was guided by the CIWA scale resulted in decreased treatment duration and total use of benzodiazepines [33].
Physical examination
On physical examination, patients typically have hepatomegaly, which often reflects the combined effects of fatty liver and swelling of hepatocytes due to cell injury-associated protein retention [34]. Signs of chronic alcohol intake (Dupuytren contracture, rhinophyma, etc.) and signs of alcohol withdrawal (tremors, tachycardia, agitation, seizures in severe alcoholic withdrawal syndrome, or delirium tremens) should be screened in primary care for early detection of ALD. Signs of chronic liver disease (spider angioma, palmar erythema and jaundice) and signs of portal hypertension (splenomegaly, ascites, and hepatic encephalopathy) suggest advanced liver disease with underlying cirrhosis. Frequently ALD patients suffer from malnutrition, physical examination may reveal sarcopenia with proximal muscle wasting and decreased grip strength protein [35].
Laboratory tests
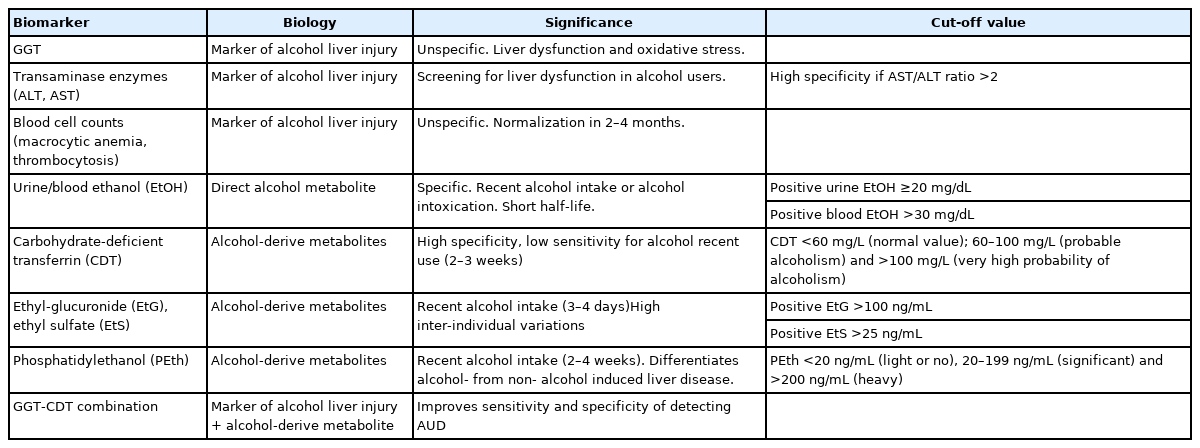
Direct alcohol biomarkers, the most used of which is ethanol detection in urine and/or blood, but they capture only very recent consumption. Alcohol metabolites are clinically used as indirect biomarkers of alcohol consumption. Metabolites of alcohol such as urine ethyl glucuronide can reveal alcohol use up to 3–4 days after the last alcohol drink. Sensitivity and specificity of urinary ethyl glucuronide for detection of alcohol use were 89% and 99%, respectively, among patients with ALD before and after liver transplant [36,37]. Measurement of ethyl glucuronide in hair samples can detect alcohol use for a longer period of up to 1 month [38]. Other metabolite of alcohol, blood phosphatidylethanol has a half-life of approximately 10–14 days, with sensitivity of 91% and specificity of 77% [39]. Carbohydrate-deficient transferrin (CDT) has a half-life of 2–3 weeks but its utility is limited by its low sensitivity of 25–50% in several studies and by false-positive results. The levels of CDT may be confounded with increasing disease severity and active smoking. Posttransplant use of %CDT appears to be more accurate, likely due to improved liver function. The CDT combined with GGT has higher sensitivity (75–90%) [40].
In laboratory test, ALD-induced liver injury indicators are also observed. Increased GGT and aspartate transaminase (AST) levels with typically AST two times greater than alanine transaminase (ALT), macrocytic anemia and thrombocytosis. According to the CLASH study, AH patients presented greater AST/ALT ratio and lower GGT compared to ASH population. In ASH patients, GGT levels were higher, probably reflecting improved preservation of hepatocyte mass (Table 1).
Genetic factors are also involved in the onset, progression, and clinical outcome of ALD. Epidemiological studies conducted among family members and between twins strongly support a genetic component [41-43]. According to the CLASH study, AH patients showed marked deregulation of genes involved in hepatocyte reprogramming and bile acid metabolism. ASH patients showed a deregulated expression of genes involved in matrisome and immune response.
It is important to consider that in the pathogenesis of liver diseases frequently coexist two different risk factors for liver injury in the same subject, potentially increasing the risk and severity of liver damage. Alcohol is a frequent co-factor in patients with hepatitis C virus infection where it accelerates hepatic fibrosis [44,45]. Additionally, alcohol has a synergistic hepatotoxic effect with nonalcoholic fatty liver disease, iron overload and other metabolic disorders [6,46-49].
Non-invasive diagnosis
As mentioned previously, fibrosis is one of the main prognostic factors in ALD. As liver biopsy is not always available, non-invasive methods to assess fibrosis have been developed (Tables 2, 3) [3]. Some alternatives are serum biomarkers, such as Fibrotest®, Fibrometer®, Hepascore® and Enhanced Liver Fibrosis (ELF) test [50]. The diagnostic accuracy of these tests is greater than other biomarkers developed for viral hepatitis (i.e., AST to Platelet Ratio Index, Forns index, Fibrosis-4 Index). These simple serum-based parameters obtained from routine liver function tests can distinguish between patients with no fibrosis or advanced fibrosis, but they have limited value to assessed intermediate stages of fibrosis. A combination of any of these tests has not been useful in improving diagnostic performance [51].
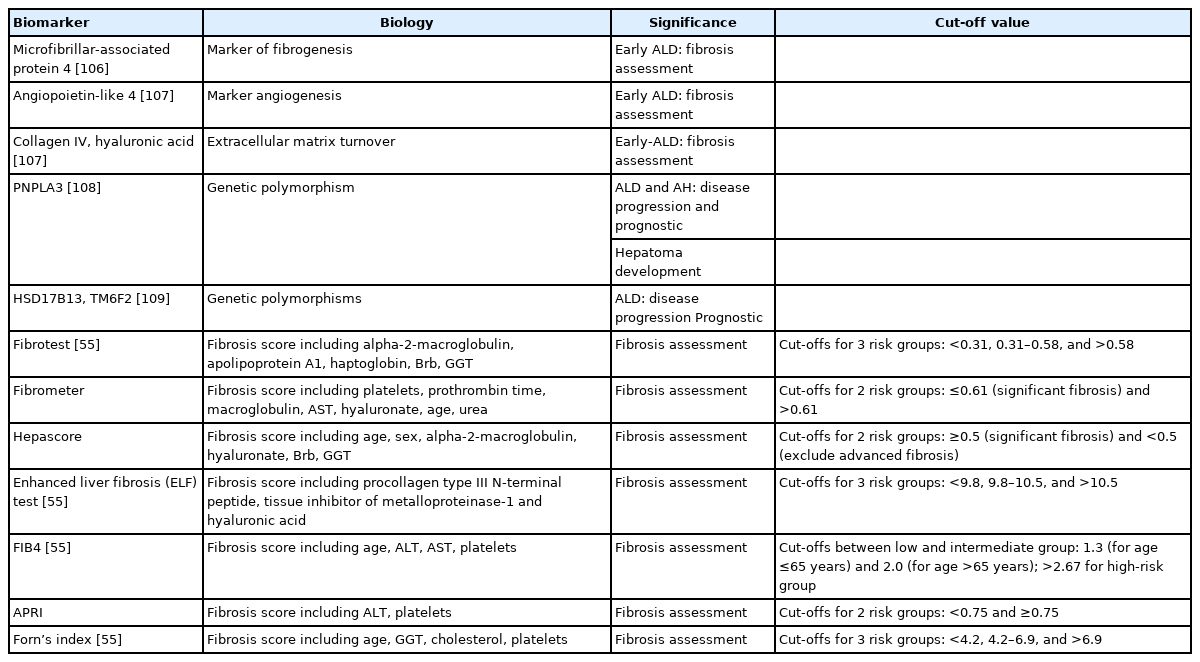

Biomarkers in diagnosis and prognosis of patients with AH
Imaging biomarkers are based on the evaluation of the liver parenchyma stiffness. Elastography measures are based on the speed and elastic wavelength that propagates through the liver tissue. As the stiffness of the liver parenchyma increases related to fibrosis, the elastography value also increase. There are different methods of generating ultrasound/elastography waves such as transient elastography and magnetic resonance elastography. Transient elastography could potentially diagnose subclinical liver disease among heavy drinkers, allowing for earlier referral to a specialty liver clinic. Liver stiffness measurement by transient elastography (FibroScan®) closely correlates with the degree of fibrosis, but in studies that did not consider the presence of AH as a possible confounder. Patients with alcohol-related cirrhosis had significantly higher values of liver stiffness than those with viral cirrhosis. Moreover, the cut-off values should be modified in patients with elevated transaminases (AST >200 UI/L) [52]. Other confounding factors that can interfere include recent alcohol intake, no compliance with fasting recommendation before the assessment and the presence of cholestasis [53]. Also, a new measurement, the controlled attenuation parameter incorporated into the FibroScan®-device showed a good correlation with steatosis on liver histology [54]. In a prospective, single-etiology cohort study of 462 patients with biopsy-proven ALD and up to 7 years of follow-up, it was found that transient elastography and the ELF test predict liver-related events with excellent prognostic accuracy. They were considered as accurate prognostic markers in patients with early stages of alcohol-related liver fibrosis or compensated cirrhosis. This study also found that cut-offs for transient elastography can be used to separate patients into three groups of distinctly different risks profiles: compared to patients with a liver stiffness below 10 kPa, patients with liver stiffness between 10 and 15 kPa had an 8-fold higher hazard for liver-related events, and those with liver stiffness >15 kPa had a 28-fold higher hazard [55]. In the near future, our group will start an observational study to identify the prevalence of advanced liver fibrosis among patients with excessive alcohol intake using a non-invasive method (transient elastography FibroScan®) and to characterize the main environmental, genetic and epigenetic factors that could influence the development of advanced fibrosis. A new tool has been implemented in conventional ultrasound systems, the shear wave elastography (SWE). It allows to choose the best acoustic window for liver stiffness measurement [56]. In a study carried out in Korea, the area under the receiver operating characteristic (AUROC) of SWE for discriminating ASH from simple steatosis was 0.93 and the AUROC for diagnosing cirrhosis with ASH vs. cirrhosis without ASH was 0.92 [57]. Additionally, a cut-off higher than 20 KPa carries prognostic information [58]. Although magnetic resonance elastography has the best diagnostic accuracy for liver fibrosis detection, its’ use is not widely available in clinical settings because it requires specific software and an external device [59,60].
A recent study carried out by Chen et al. [61] has also identified some prognostic factors based on computed tomography radiomics (texture, liver surface nodularity and steatosis measurements) which were associated with reduced 90-day and overall transplant-free survival in AH.
Histology assessment
Histologically, AH is associated with ballooned hepatocytes, MDB, lobular polymorphonuclear neutrophils and pericellular and sinusoidal fibrosis (in “chicken wire” appearance). The histological features of ASH are similar from those described in NASH [62].
According to the CLASH study, AH presented histologically more advanced fibrosis, MDB, bilirubinostasis, severe neutrophil infiltration and progenitor cell expansion than ASH. AH was characterized by profound ductular reaction, which is not seen in early asymptomatic phases and is associated with poor prognosis. Another study carried out by Ventura-Cots et al. [63] revealed through histological and molecular profiling that ductular reaction is a driver of portal hypertension in patients with AH.
The SALVE (Study of Alcohol-related LiVer disease in Europe) Histopathology Group developed and validated a grading and staging system for the clinical and full histological spectrum of ALD and evaluated its prognostic utility in a multinational cohort of 445 patients [27]. SALVE grade was described by semiquantitative scores for steatosis, activity (hepatocellular injury and lobular neutrophils) and cholestasis. The histological diagnosis of ASH due to ALD (histological ASH) was based on the presence of hepatocellular ballooning and lobular neutrophils. Severe cirrhosis and histological ASH were identified as independent predictors of short-term survival in decompensated ALD, and decompensation- free survival in compensated ALD.
Altamirano et al also presented a histologic scoring system that relates to the prognosis in patients with AH [23]. The authors identified histologic features associated with AH disease severity and proposed a semiquantitative scoring system, the “Alcoholic Hepatitis Histologic score”. Four histologic features were combined to create the final score: fibrosis stage, bilirubinostasis, polymorphonuclear infiltration, and megamitochondria. The degree of fibrosis and the presence of bilirubinostasis were positively associated with higher short-term mortality. On the other hand, mild polymorphonuclear infiltration and absence of megamitochondria were associated with poorer outcome in these patients. Low, moderate, and high score was associated with a short-term mortality of 3%, 19%, and 51%, respectively. The prognostic assessment of this score to predict short-term mortality compared favorably with well-validated, non-invasive scoring systems for AH such as the Model for End-Stage Liver Disease (MELD). Further, the histologic score was able to provide additional prognostic information in AH patients with low MELD scores. In patients with a MELD score of <21, a cutoff value of 5 points in the histologic score differentiated two subgroups with different 90-day survival (94% vs. 72%). However, a recent study showed that this score is not predictive of shortterm survival in patients with severe AH. Further studies should evaluate whether the prognostic value of histologic parameters differs according to the severity of liver disease [64].
AH
AH is clinically defined as abrupt onset of progressive jaundice and liver-related complications with hyperbilirubinemia (>3 mg/dL), AST/ALT ratio >1.5 with levels of AST >1.5 times the upper limit of normal but <400 IU/L; and heavy alcohol drinking until 4 weeks before onset of symptoms and absence of other causes of liver disease (10–20%) [65].
To date, multiple scoring systems have been developed to predict short-term prognosis in patients with AH. One of the most validated is the Maddrey Discriminant Function (MDF), which includes prothrombin time and total bilirubin, and where severe AH is defined by a score ≥32 [66]. Other scoring systems include the MELD score [67], the Glasgow Alcoholic Hepatitis score [68] and the age, serum bilirubin, INR and serum creatinine [69,70]. MELD score incorporates renal function, as a major determinant of outcomes in AH patients. A MELD score >20 has been proposed as definition of severe AH [71]. A recent large worldwide study showed that MELD is the best scoring system to predict mortality in alcohol-associated hepatitis [72]. Another relevant prognostic tool is the Lille score, assessed after 4–7 days of corticosteroid therapy. A Lille score <0.45 predicts a good response to corticosteroids and continuing prednisolone for 4 weeks is recommended [73].
Acute kidney injury (AKI) is a common complication in patients with AH and negatively impacts short-term survival [74,75]. Therefore, serum creatinine should be screened in all patients with AH. A recent study carried out by Fernandez-Carrillo et al. [76] found out that AKI in the setting of AH is characterized by a higher urine potassium concentration. Among the biomarkers considered in the study, urine NGAL, IGFBP-7, KIM-1, LFABP, as well as TIMP-1 and TIMP-2 discriminated AKI vs. non-AKI (P<0.01 for all). Interestingly, the performance of TIMP-1 and 2 in patients with AH was significantly better than in patients with decompensated cirrhosis with and without AKI. Interestingly, urine interleukin (IL)-18 was exclusively increased in patients with AH and identified those with AKI (P<0.001). Serum levels of IL-18 were slightly higher in patients with AH vs. decompensated cirrhosis (P<0.05). There was no correlation between serum and urine IL-18 levels among patients with AH. Among all studied biomarkers, NGAL predicted 3-months survival (AUROC 0.72) [76]. Hepatic encephalopathy and lack of alcohol abstinence are also key factors that impair long-term prognosis [77].
Patients with non-severe AH, defined as a MELD score ≤20 or MDF <32, have a low risk of short-term mortality. The 5-year mortality of decompensated patients with ASH and an MDF <32 is about 50%.
Regarding the AH treatment, the only therapy with proven benefit is alcohol abstinence. There are multiple scores to predict alcohol relapse after an AH episode. A recent study carried out by Clemente et al. [78], discriminate different risk groups for early alcohol relapse after an episode of AH. The higher group risk were patients under 44 years of age, MELD >21, no cirrhosis or psychiatric disease diagnosis, no relationships and unemployed [78]. A therapy that has shown likely benefit is corticosteroids. Prednisolone (40 mg/day) given orally should be considered to improve 28-day mortality in patients with severe AH (MDF ≥32) without contraindications to the use of corticosteroids. However, survival benefit was not sustained at 90 or 180 days [79]. The Lille score should be used to reassess prognosis, identify non-responders, and guide treatment course after 7 days of corticosteroids [80]. Other therapies with potential benefit are N-acetylcysteine and Granulocyte-colony stimulating factor (G-CSF) [81]. In patients with steroid non-responsive severe AH (day 7 Lille score >0.45), the administration of G-CSF reduces the disease severity and 90-day mortality [82]. According to a recent presentation at the International Liver Congress 2021 carried out by Louvet et al. [83], amoxicillin/clavulanate plus prednisolone in severe AH do not improve survival at 2 months. Future treatment to consider are IL-1R antagonist anakinra, fecal transplantation [84], DUR-928 [85], and IL-22 agonist F-652 [86]. It should also be considered the early liver transplant for severe AH not responding to medical therapy [14], it has been demonstrated that improves survival compared to patients without transplant [87-90].
SUBCLINICAL ASH VS. AH: MOLECULAR PROFILING
The cellular and molecular mechanisms of ALD are multifaceted, complex, and poorly understood in part due to the lack of clinical and histological replication in animal models. The majority of mechanistic investigations uses animal models to identify several molecular drivers of intermediate ALD (steatosis and mild inflammation), but not the severe fibrosis and cholestasis that are seen in advanced ALD [6].
Alcohol is processed into acetaldehyde in the liver forming proteins and DNA adducts, promoting lipid peroxidation, glutathione depletion, and mitochondrial damage [91]. These adducts also act as antigens, causing lymphocyte migration to the liver and activating the adaptive immune response. Kupffer cells release anti-inflammatory cytokines (IL-10) and hepato-protective factors (IL-6) that help to prevent hepatocellular damage caused by alcohol [92,93].
On top of that, acute alcohol intake or binge drinking also increase serum levels of bacterial products. Alcohol increases gut permeability and bacterial product translocation into the portal circulation, leading to the generation of proinflammatory cytokines such tumor necrosis factor, which contribute to hepatocellular damage [94]. A variety of proinflammatory cytokines, such as IL-1, IL-8, osteopontin, CXCL1, CXCL4, CXCL5, and CXCL6, are up-regulated during alcoholic liver injury and also contribute to neutrophil recruitment [24,95]. Recently, there is an increasing amount of evidence suggesting the gut-liver axis and bacterial dysbiosis as a major factor in ALD, potentially becoming a target for therapy. There are major imbalances in the gut barrier that facilitates pathogen-associated molecular patterns, gut-derived bacterial products that trigger mostly hepatic stellate cells (HSCs) and Kupffer cells, to translocate into the portal circulation [96]. The more severe the AH onset gets, the more gut dysbiosis and bile-acid disbalance is seen [94].
The major pathogenic and prognostic event in the development of ALD is the progression of fibrosis [97-99]. Alcohol abuse can cause liver fibrosis by extracellular buildup of collagen and other matrix proteins. It promotes collagen expression in HSCs and, when coupled with other biological components, forms a variety of adducts that keep HSCs active [99]. Neutrophils, injured hepatocytes, and activated Kupffer cells can also stimulate HSCs by releasing profibrogenic mediators such as transforming growth factor, platelet-derived growth factor, IL-8, angiotensin II, and leptin [97]. HSC profibrogenic signaling pathways are also stimulated by reactive oxygen species, resulting in fibrogenesis enhancement through modulation of angiogenesis. The endoplasmic reticulum and mitochondrial-derived glutathione continuously neutralize the reactive oxygen species produced by the metabolism of alcohol. However, whenever the body is exposed chronically to alcohol, mitochondrial-derived glutathione becomes depleted, and the reactive oxygen species interact with iron and ethanol forming reactive metabolites responsible for lipid peroxidation of cell membranes [100,101].
Activated HSCs are destroyed by natural killer cells, which release interferon, cause HSC cell cycle arrest and apoptosis [102,103]. Thus, alcohol reduces the ability of natural killer cells to fight fibrosis and liver regeneration becomes inefficient. In advanced cirrhotic ALD, chronic alcohol exposure impairs liver regeneration by inhibiting DNA synthesis in mature hepatocytes. Thus, along with hepatocyte dedifferentiation, the liver regeneration impairment is a key event leading to liver failure in ALD [6].
The proliferation mechanisms of adult hepatocyte are entirely substituted by massive ductular cell proliferation and an intensification in the number of hepatocytes expressing markers from progenitor cell [2]. Our group has shown recently that patients with AH have a hepatocyte nuclear factor 4 alpha (HNF4A) transcriptomic footprint. Liver-enriched transcription factors, such as HNF1A, RXRA, and FOXA1, were shown to be downregulated, while HNF4A P2 variants, characteristically expressed throughout fetal liver development, are expressed in AH patients [104]. Thus, the restoration of HNF4A function can potentially be a therapeutic target for severe ALD (Fig. 3).
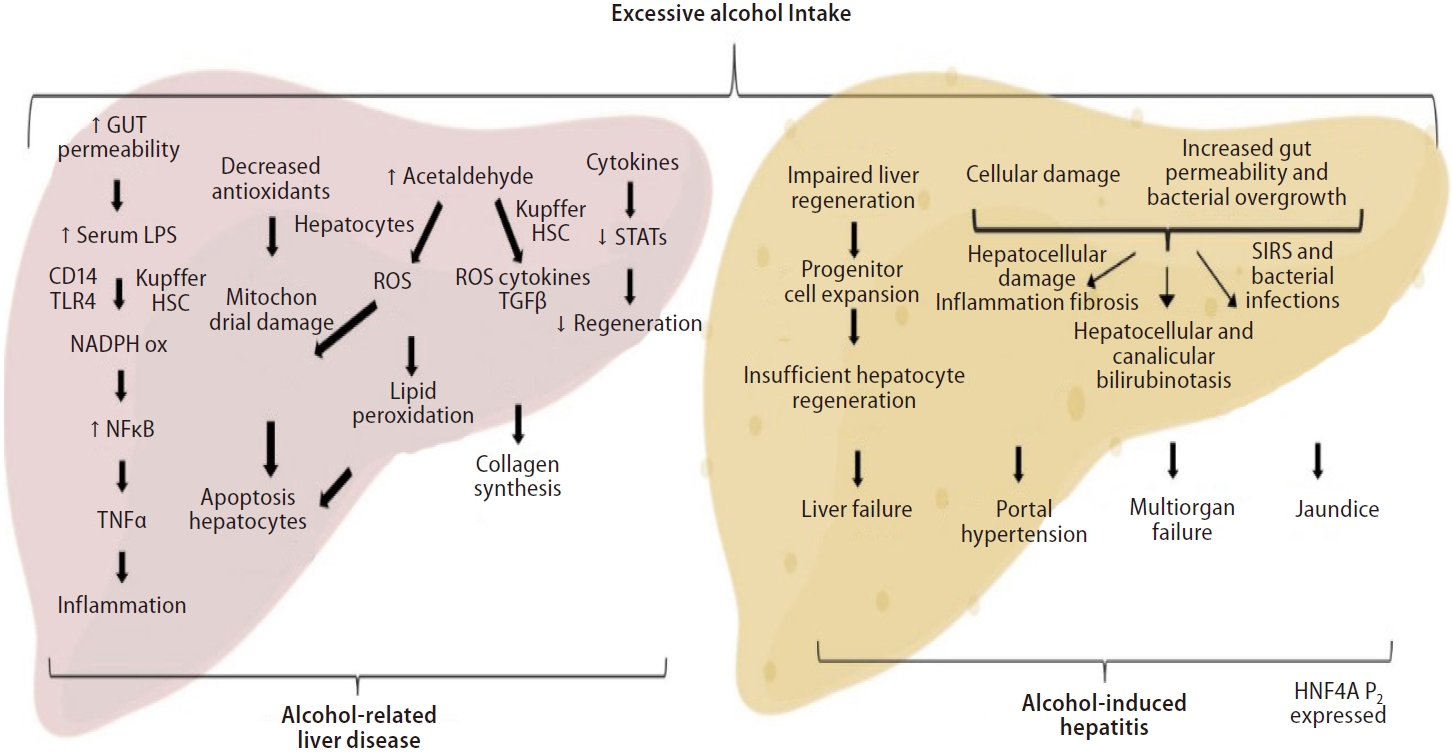
Pathogenesis of alcohol-related liver disease and alcohol-induced hepatitis. Excessive alcohol intake cause Kupffer cells and Stellate cells activation along with changes in the microbiome and increased gut permeability. The resulting liver damage, hepatic and systemic inflammation and liver fibrosis are responsible for the dominant clinical consequences seen in AH patients. In addition, expansion of immature progenitor cells leads to impaired regeneration and HNF4a P2 variants are seen expressed in AH. LPS, lipopolysaccharide; NADPH, nicotinamide adenine dinucleotide phosphate; NFκB, nuclear factor kappa B; TNF, tumoral necrosis factor; ROS, reactive oxygen species; HSC, hepatic stellate cell; TGF-β, transforming growth factor-β; STAT, signal transducers and activators of transcription; SIRS, systemic inflammatory response syndrome; HNF4A, hepatocyte nuclear factor 4 alpha; AH, alcohol-induced hepatitis.
CONCLUSIONS
The prevalence of AUD and ALD is increasing in a global manner. ALD includes a wide range spectrum of early and advanced phenotypes. Most patients are seen at advanced stages of the disease when an episode of AH and/or clinical decompensations are developed. Campaigns for early detection of asymptomatic or subclinical forms are urgently needed. ALD is characterized by profound fibrogenesis even in subclinical forms. Compared to compensated ALD, AH is characterized by profound reprogramming of hepatocytes with features of de-differentiated and cholangiocytes. Translational studies have identified novel targets for the design of therapeutic clinical trials.
Unmet needs for future research
Scientific consensus conference to better define subclinical stages of ALD. Cost-effective measures and public health policies to reduce alcohol consumption. Early detection of AUD and concomitant ALD is urgently needed. As the underlying mechanisms of subclinical ASH remain elusive, further research is needed to clarify druggable molecular drivers as well as prognostic biomarkers. Effective and safer therapies for patients with ALD are still needed.
Notes
Authors’ contribution
CGM and LM wrote the manuscript and prepared the figures and tables. DMA and RB revised the manuscript. All the authors read and approved the final version.
Conflicts of Interest
The authors have no conflicts to disclose.
Abbreviations
ACLF
acute-on-chronic liver failure
AH
alcohol-induced hepatitis
AKI
acute kidney injury
ALD
alcohol-related liver disease
ALT
alanine transaminase
ASH
steatohepatitis
AST
aspartate transaminase
AUD
alcoholic use disorder
AUDIT
Alcohol Use Disorders Identification Test
AUROC
area under the receiver operating characteristic
CDT
carbohydrate-deficient transferrin
CIWA
Clinical Institute Withdrawal Assessment for Alcohol
DALYs
disability-adjusted life years
ELF
Enhanced Liver Fibrosis
G-CSF
granulocyte-colony stimulating factor
GGT
gamma-glutamyl transferase
HNF4A
hepatocyte nuclear factor 4 alpha
HSCs
hepatic stellate cells
IL
interleukin
MDB
Mallory-Denk bodies
MDF
Maddrey Discriminant Function
MELD
Model for End-Stage Liver Disease
NAFLD
non-alcoholic fatty liver disease
SALVE
Study of Alcohol-related LiVer disease in Europe
SWE
shear wave elastography