INTRODUCTION
The introduction of targeted therapy for cancer treatment increased demand for identification of molecular targets, such as driver mutations in cancer cells [1]. However, many molecular tests, including next-generation sequencing, are not available to all cancer patients because of high cost and technical barriers. Recently, deep learning (DL) has been applied to predict molecular biomarkers directly from Hematoxylin and Eosin (H&E)-stained cancer tissue slides [2]. After introduction of high-speed digital slide scanners and the recent US Food and Drug Administration approval of digitized whole slide images (WSIs) as primary specimens for diagnosis, digitization of tissue slides has been widely adopted in clinical practice [3]. Accumulation of digitized tissue images enables DL model training for prediction of many molecular pathology test results, such as mutational status of driver genes and microsatellite instability (MSI). Furthermore, the response to specific anticancer agents and the prognosis of cancer patients can be predicted by DL. Since H&E-stained tissue slides are prepared for almost all cancer patients, the DL-based method can be a cost- and time-effective alternative tool for clinical decision making [4]. In this review, we explain the current state of DL applications for the prediction of genetic mutation, MSI, tumor mutational burden (TMB), molecular subtypes, gene expression, treatment response, and prognosis directly from H&E-stained tissue slides.
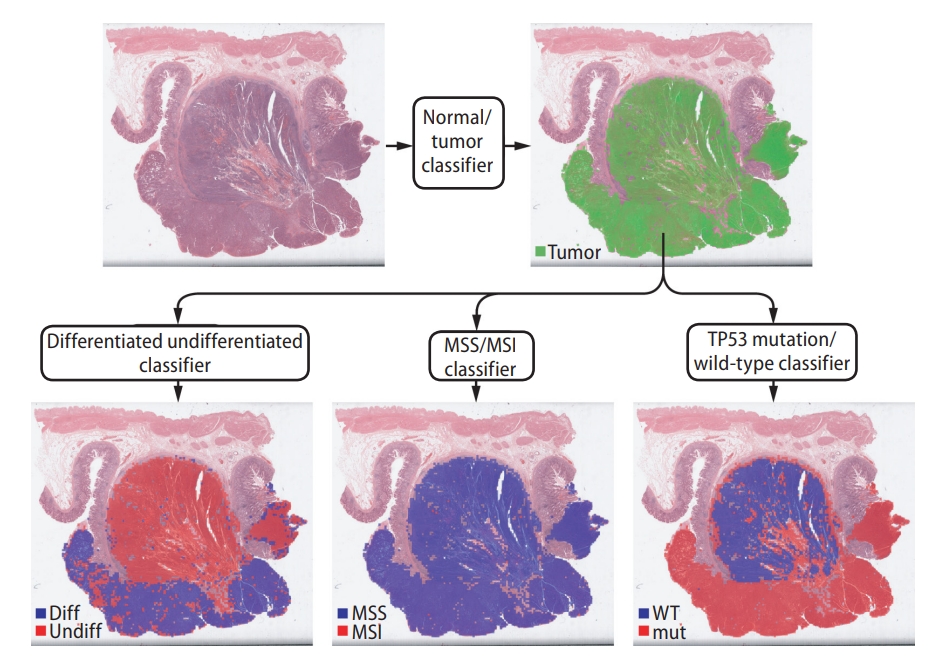
Because this review is written for cancer researchers and clinicians who are not familiar with DL, we would like to start with an explanation of the basic procedure to process the WSIs of tissue slides with DL to classify tissues into different types. As an example, Figure 1 shows how normal and tumor tissue classifiers can be trained. The left image in panel A shows a WSI with regions of normal and tumor tissues labeled by pathologists. The labeled regions in the WSI are split into small image patches and collected for each label, as demonstrated in the middle part of panel A. The split is inevitable because a WSI is usually a very large image with a size of 100,000├Ś100,000 pixels when scanned at 40├Ś magnification. The current state of DL systems does not allow such large images to be processed at once. Supplementary Figure 1 presents examples of 250├Ś250 pixels tissue image patches at different magnifications. Oftentimes hundreds to thousands of WSIs must be collected for accurate DL models, and the total number of collected image patches sometimes exceeds one million for each class. DL is a process to train a deep neural network (DNN) to yield correct classification results. When the task is image classification, a specific DNN called a convolutional neural network (CNN) is used. Therefore, image patches are presented as inputs to the CNN, and the outputs of the CNN are processed to yield results. Then, the classification results are compared with the true labels of the images, and the CNN is iteratively modified to yield correct results. It is common for the CNN initially to fail to correctly classify images. As shown in the first image of Figure 1B, the CNN classifies all image patches as tumors at the beginning of training, which is incorrect. As training progresses, the network yields a larger proportion of correct results because the mathematical weights used to calculate prediction are adjusted based on incorrect results. The last image of panel B is the classification results for an entire WSI after 20 hours of training. However, different from tumor classification, many molecular biomarkers inside the WSI are not identified and labeled by pathologists because it is unclear which regions show the biomarker-specific features. Therefore, WSIs are usually labeled as entirely negative or positive for specific biomarkers, and all image patches from a WSI have the same label. An example of this scenario regarding prediction of MSI status is presented in Supplementary Figure 2.
Published papers on the prediction of genetic mutation, MSI, TMB, molecular subtype, gene expression, treatment response, and prognosis are summarized in the following sections. Training and external validation cohorts are important for evaluating the results of such studies. Most studies adopted molecular test results and tissue slides provided by The Cancer Genome Atlas (TCGA) for either training or validation. The type of CNN and the size of image patches are also important in the DL-based evaluation of tissue images. Finally, performance measures should be compared between studies; most studies defined the DL-based model performance as the area under the receiver operating characteristics curve (AUROC). These key features of the reviewed papers are summarized in tables.
GENETIC MUTATION
An increasing number of drugs targeting specific mutations in cancer cells has been developed. For example, gefitinib and erlotinib are effective against non-small cell lung cancers with EGFR mutation [5]. Vemurafenib is effective against BRAF-mutated metastatic colorectal cancer (CRC) [6], and Sotorasib selectively and irreversibly targets KRASG12C in solid cancers [7]. Mutational status should be assessed by various sequencing or staining methods to select patients for these targeted drugs. Additional cost, time, and tissue samples are required for these tests. Therefore, not every patient can be properly tested for optimal treatment options. Many studies have tried to predict the mutational status of various genes from H&E-stained diagnostic tissue slides of multiple cancer types to select patients for these targeted drugs. Table 1 summarizes the results of the studies.
One of the first studies demonstrating the feasibility of DL-based mutation prediction for H&E-stained tissue slides was published on bioRxiv in 2016. Schaumberg et al. [8] tried to predict the mutational status of the SPOP gene from prostate cancer tissue slides provided by TCGA. The model performed well with the external validation cohort (MSK-IMPACT), demonstrating the feasibility of DL-based mutation prediction.
In 2018, seminal work for DL-based prediction of mutational status of multiple genes directly from H&E-stained histopathologic images of lung cancer was published by Coudray et al. [9]. The authors first tested whether DL could discriminate between adenocarcinoma and squamous cell carcinoma of the lung. Then, they focused on adenocarcinoma for determination of the 10 most frequently mutated genes. DL models could discriminate the mutational status of STK11, KRAS, SETBP1, EGFR, FAT1, and TP53 with the AUROCs of 0.845, 0.814, 0.785, 0.754, 0.739, and 0.674, respectively.
Because the mutational status of BRAF and NRAS is important for therapeutic decision-making regarding those with stage III/IV melanoma, Kim et al. [10] built prediction models for BRAF and NRAS mutations. Interestingly, predictive performance was affected by the thickness of the tumor and ulceration status. In skin cancer, these parameters reflect complex underlying molecular mechanisms that control the phenotype of mutated genes. Therefore, independent models considering different thicknesses and ulceration statuses can improve the overall performance of mutation prediction in melanoma.
Tsou and Wu tried to discriminate two mutually exclusive drivers of papillary thyroid carcinoma: BRAFV600E and mutated RAS [11]. The accuracies of the classifiers were 63.6% and 90% for BRAF and RAS mutations, respectively, with an overall AUROC of 0.951 for the test set.
Differentiation status and mutations were predicted by Chen et al. [12] from liver cancer tissue slides. The authors first tried to classify the liver cancer tissues into well, moderately, and poorly differentiated tumors and compared the results with pathologists. Then, they predicted the mutational status of the 10 most commonly mutated genes. Four of them, CTNNB1, FMN2, TP53, and ZFX4, were predicted with the AUROCs ranging from 0.71 to 0.89.
Because the mutational statuses of IDH genes (IDH1 and IDH2) are important prognostic and therapeutic biomarkers for glioma, Liu et al. [13] tested the feasibility of IDH mutation prediction from histopathologic glioma images using DL. They tried to augment existing images using a Generative Adversarial Network (GAN). GANs can create virtual images of glioma with and without IDH mutation by iteratively improving generated images until they are indiscernible from the training set. Using a GAN, the AUROC was slightly improved from 0.920 to 0.927. Although there was some improvement, these results indicated that generative modeling could provide only a slight improvement of performance because GAN-created images are only extended virtual copies of existing images and cannot provide new information.
Fu et al. [14] tried to build a general-purpose classifier for multiple mutations in various cancers. They first constructed an Inception v4-based classifier for discrimination among 42 tissue types, including 28 cancers and 14 normal tissues. Then, the classifier was reused to predict the mutational status of multiple genes. In detail, regression methods were applied to the 1,536 features from the final layer of the classifier for cancer type-specific mutation prediction. The AUROCs ranged from 0.098 for the FBXW7 gene in head and neck cancer to 0.972 for the STAG2 gene in bladder cancer. The performance seemed suboptimal because an end-to-end model was not used for mutation prediction; however, the possibility of a pan-cancer classifier was well demonstrated.
Kather et al. [15] published another pan-cancer approach for detection of clinically actionable genetic alterations. They aimed to predict mutations affecting at least four patients, a prevalence above 2%. The results were suboptimal compared to those of other studies that investigated specific cancer types. For example, the AUROCs for APC, KRAS, SMAD4, and TP53 mutations in CRC were 0.63, 0.6, 0.61, and 0.64, respectively, which were much lower than those of our previous study, 0.771, 0.778, 0.693, and 0.809 [16].
Noorbakhsh et al. [17] trained classifiers to predict TP53 gene mutational status from breast, lung, stomach, colon, and bladder cancers from the TCGA datasets and obtained the AUROCs of 0.7447, 0.7969, 0.6532, 0.7825, and 0.7094, respectively. Importantly, they found that the classifiers could identify mutational status more accurately when tumor tissues were more homogenous. As we discuss later, tumor heterogeneity affects the performance of DL-based tissue classifiers.
In our previous study, mutations in APC, KRAS, PIK3CA, SMAD4, and TP53 genes were predicted from CRC tissue slides [16]. We trained classifiers for frozen and formalin-fixed paraffin-embedded (FFPE) tissues separately because they are morphologically different. We previously showed that DL-based classifiers are incompatible between frozen and FFPE tissues [18]. The AUROCs ranged from 0.693 to 0.809 and from 0.645 to 0.783 for frozen and FFPE tissues, respectively. When we combined the TCGA data and our own data for the training of the models, the performance was improved. These results indicate that collection of larger dataset is essential to improve the performance of DL-based classifiers.
Yang et al. [19] investigated the feasibility of mutational status prediction for DNMT3A, EGFR, PBRM1, STK11, and TP53 genes in lung cancer, which are thought to be candidate markers for immunotherapy response. The AUROCs were between 0.71 and 0.87.
Because fibroblast growth factor receptor (FGFR) inhibitor was approved as a targeted therapy in bladder cancer, Loeffler et al. [20] tried to predict the mutational status of the FGFR gene from tissues of bladder cancer. Furthermore, they compared the performance of the DL model with that of pathologists because there are some visually recognizable histologic features in FGFR-mutated tumors. The AUROC of the DL model was 0.701, and it outperformed pathologists. Interestingly, one tissue had available information on intratumor heterogeneity, which was investigated with multi-region sequencing. DL correctly delineated the heterogeneous regions in the tissue.
We also tried to predict mutations in gastric cancer (GC) tissue slides [21]. Similar to the study for the CRC, we separately trained the classifiers for frozen and FFPE tissues and predicted the mutational status of CDH1, ERBB2, KRAS, PIK3CA, and TP53 genes. The AUROCs ranged from 0.727 to 0.862 and from 0.661 to 0.858 for frozen and FFPE tissues, respectively. The performance could also be enhanced by combining the TCGA data with our own data, highlighting the importance of large datasets for prediction of mutational status from H&E-stained tissue slides.
MSI
Accumulation of insertions or deletions in the repeated units of microsatellites by impaired DNA mismatch repair (MMR) causes MSI [22]. Deficient MMR also increases overall mutation rates and leads to expression of neoantigens, which attract immune cells. To avoid immune surveillance, tumor cells express several immune checkpoint ligands [23]. Therefore, MSI could be a marker for patient response to immune checkpoint inhibitors (ICIs) [24]. Furthermore, MSI is an important prognostic marker [25]. Many studies tested the feasibility of MSI status prediction from tissue slides using DL (Table 2).
The first comprehensive study to investigate the feasibility of DL-based MSI status prediction was published by Kather et al. [26] in 2019. They focused on gastrointestinal (GI) cancers, and GC and CRC datasets from the TCGA program were used to train the classifiers. The AUROCs were 0.81 and 0.77 for the FFPE tissue slides of GC and CRC, respectively, and 0.84 for the frozen tissue slides of CRC. When the DL models were tested on an Asian GC cohort and the TCGA endometrial cancer dataset, performance was much decreased. These results indicated that the classifiers did not extend beyond the ethnicity and cancer types presented in the training datasets. Therefore, current generalizability of DL-based classifiers for MSI status appears to be limited.
Cao et al. [27] also tested DL-based MSI status prediction in CRC. The AUROC was 0.8848 for frozen tissue slides from the TCGA. When the classifier was tested on an Asian CRC cohort, the AUROC was only 0.6497. The authors implemented the transfer learning scheme to improve the performance on the Asian-CRC cohort. When the parameters of the classifier trained on the TCGA dataset were transferred to retrain a new classifier for the Asian-CRC cohort, the AUROC was improved to 0.8504. These results confirmed that transfer learning is a plausible method to modify a classifier to improve performance on other datasets.
Echle et al. [28] formed the MSIDETECT consortium to collect a large set of CRC data and improve the performance of MSI prediction. They recruited more than 8,000 patients and improved the AUROC to 0.92, which is much better than their previous TCGA-based study with the AUROCs between 0.77 and 0.84 [26]. Although they clearly demonstrated the importance of a large dataset for better performance, the importance of ethnicity on predictive performance was not tested because there was no Asian cohort involved.
Wang et al. [29] trained a prediction model for MSI status in uterine corpus endometrial carcinoma. The AUROC of their model was 0.73, not superior to that of Kather et al. [26].
Similar to the study by Liu et al. [13], Krause et al. [30] trained a GAN to enlarge the tissue datasets for MSI research by generating virtual tissue images. The use of GAN-created images improved the AUROC from 0.757 to 0.777. Compared to the study by Liu et al. [13], this improvement was more prominent.
Yamashita et al. [31] compared the performance of a DL-based MSI status prediction model with that of five GI pathologists. The pathologists discriminated the MSI tissues based on 10 known tissue features of MSI CRC tumors, such as CrohnŌĆÖs-like reaction and signet ring-cell differentiation. Regarding the reader experiment (40 cases), the AUROC of the DL model was 0.865. The mean AUROC for the five pathologists was 0.605. Therefore, the DL model outperformed the pathologists.
We also tested the feasibility of DL-based MSI status prediction and obtained the AUROCs of 0.942 and 0.861 for frozen and FFPE tissues, respectively, from the TCGA CRC datasets [32]. The classification performance was not satisfactory on our own Asian dataset, with an AUROC of 0.787. This result reiterated the lack of compatibility of TCGA-based MSI prediction models on Asian cohort. Then, we trained a new classifier with both TCGA and our own dataset. The new classifier performed well on both datasets, with the AUROCs of 0.892 and 0.972 for the TCGA and our dataset, respectively. These results demonstrated the importance of large multi-national datasets for better performance. We also showed that the application of a classifier trained on the CRC tissues from original sites could not be extended to metastasized CRC tissues in the liver and lung (AUROC, 0.484). These results indicated that the morphologic features of MSI in tumor tissues were different between primary and metastasized tumors. We also showed that the DL model discriminated MSI tissue images based on previously known features, such as cribriform pattern, high tumor-infiltrating lymphocyte (TIL) density, and mucinous differentiation. Because these features were more prominent in primary tumors than metastasized tumors, the DL algorithm could discriminate MSI more clearly in primary tumors.
TMB
TMB is defined as the total number of somatic mutations in the coding area of a tumor genome [33]. Immune cells infiltrate because of high neoantigen expression in tumors with high TMB; thus, tumor cells express several immune checkpoint ligands to avoid immune surveillance. Therefore, like MSI, TMB can be a clinical biomarker for response to ICIs [34]. TMB has been predicted in many studies (Table 3).
One of the first studies to predict TMB status was published by Xu et al. [35] in 2019. They trained a DL model to discriminate high vs. low TMB in bladder cancer and obtained an AUROC of 0.75. They also showed that the probability of survival was higher for patients with tissues predicted to have high TMB by the classifier.
Jain and Massoud [36] predicted TMB status by integrating three DL models with lung adenocarcinoma images at different magnifications (5├Ś, 10├Ś, and 20├Ś). Although the AUROCs were only 0.72, 0.80, and 0.81 for 5├Ś, 10├Ś, and 20├Ś models, respectively, the combined model achieved an AUROC of 0.92.
Another study by Xu et al. [35] extended their previous study and predicted TMB status from bladder and lung cancers, with the AUROCs of 0.752 and 0.742, respectively [37]. Most importantly, they analyzed intratumoral heterogeneity with DL and discovered better prognoses for tumors with high TMB and low intratumoral heterogeneity than for highly heterogenous high-TMB tumors. Only DL-based approaches can obtain information on intratumoral heterogeneity with affordable cost.
Shimada et al. [38] first tried to discriminate high TMB from CRC tissue slides based on TIL counts and achieved an AUROC of 0.910. Then, a DL-based model was trained and yielded an AUROC of 0.934. Although the improvement was not large, DL can achieve better results without the laborious process of TIL counting.
Sadhwani et al. [39] tried a histologic subtype-based approach to develop an interpretable model for TMB prediction. They trained a DL model to discriminate nine histologic features from lung adenocarcinoma tissues. Then, the information on tissue features was combined with clinical data to predict TMB status. Although this approach can help to improve the interpretability of DL models, the overall performance was inferior to that of a previous study [36].
MOLECULAR SUBTYPES
Cancers can be subclassified into molecular subtypes based on molecular pattern [40-43]. Molecular subtypes provide important information for clinical decision-making because they show different prognoses and therapeutic responses. However, molecular subtyping is costly and technically difficult. Therefore, DL-based prediction of molecular subtypes can be a cost-effective tool for patient stratification. Recently, the molecular subtypes of various cancers have been predicted by DL from tissue images (Table 4).
Couture et al. [44] tried to discriminate the intrinsic molecular subtypes of breast cancer. Based on the subtypes discriminated by the Prediction Analysis of Microarray 50 (PAM50), they trained a classifier to discriminate basal-like vs. non-basal-like (including luminal A, luminal B, and HER2-enriched subtypes) tumors from tissue slides. The accuracy of the molecular subtype classifier was 77%. Because they only tried to discriminate one subtype from three others, the overall performance of the DL system on classification of the four molecular subtypes in breast cancer could not be estimated.
Based on tissue type classifiers, Kather et al. [15] tried to discriminate the intrinsic molecular subtypes for breast, colorectal, gastric, and lung cancers. Basal-like, Her2-enriched, luminal A, and luminal B subtypes of breast cancer were detectable with the AUROCs between 0.61 and 0.86. For CRC and GC, the AUROCs for the pan-GI subtypes, including GI-hypermutated indel, GI genome stable, GI-chromosomally unstable, GI-hypermutated-single-nucleotide variant predominant, and GI Epstein-Barr-virus-positive, ranged from 0.23 to 0.78. TCGA molecular subtypes LUAD1 to 6 in lung adenocarcinoma yielded an AUROC of up to 0.74. These results indicate that tissue morphology reflects the characteristics of the intrinsic molecular subtypes.
Another study regarding PAM50-based molecular subtype discrimination in breast cancer was published by Jaber et al. [45]. The classification accuracy for the four classes (basal-like, Her2-enriched, luminal A, and luminal B) was 67.27%. Because there was an imbalance between the number of classes, they tried to distinguish two classes (basal-like and nonbasal-like), and the accuracy was 87.21%. Therefore, it is possible for the performance of DL models to be improved if large amounts of data on Her2-enriched, luminal A, and luminal B cases are collected. They also showed that patients with heterogeneous cancer tissues with mixed basal-like and luminal A characteristics had intermediate survival compared to homogenous basal-like and luminal A groups, suggesting the importance of tumor heterogeneity for prognosis.
Hong et al. [46] first trained a classifier to discriminate the histologic types of endometrial cancer into endometrioid or serous subtypes and achieved an AUROC of 0.969. Then, they tried to discriminate the four molecular subtypes: POLE ultramutated, high level of MSI (MSI-H) hypermutated, copynumber variation low (CNV-L), and copy-number variation high (CNV-H). The AUROCs for CNV-H and MSI-H were 0.934 and 0.827, respectively.
Four consensus molecular subtypes (CMS1 to 4) of CRC were classified by Sirinukunwattana et al. [47] from tissue images. They adopted the domain adversarial training scheme, which enforces the DL model not to learn specific features confined to a specific cohort. Therefore, the network could learn more general features. The AUROCs for CMS1 to 4 were 0.86, 0.91, 0.92, and 0.89, respectively, yielding a macro-average AUROC of 0.9.
Yu et al. [48] trained classifiers to discriminate normal and tumor tissues from non-small cell lung cancer tissues and further classified the tumor tissues into adenocarcinoma and squamous cell carcinoma. Based on the classification results, the three transcriptomic subtypes of lung adenocarcinoma, terminal respiratory unit, proximal inflammatory, and proximal proliferative subtypes, and the four transcriptomic subtypes of lung squamous cell carcinoma, classical, basal, secretory, and primitive subtypes, were classified. The AUROCs for the adenocarcinoma subtypes ranged from 0.771 to 0.892, and the AUROCs for the squamous cell carcinoma subtypes were around 0.7.
Based on a custom panel of 21 genes, Woerl et al. [49] predicted four molecular subtypes of muscle invasive bladder cancer. The AUROCs for discrimination of double negative, basal, luminal, and luminal p53-like subtypes were 0.76, 0.89, 0.88, and 0.89, respectively. They found that DL-based prediction outperformed morphology-based prediction by four pathologists.
EXPRESSION OF GENES AND PROTEINS
Expression of specific genes and proteins has a significant impact on prognostication and therapeutic decision-making [50-53]. DL has been successfully applied for prediction of expression status, particularly for breast and lung cancers (Table 5).
Estrogen receptor (ER) expression has significant implications in the prognosis and treatment of breast cancer [54]. Couture et al. [44] trained a DL model to test the ER status of breast cancer tissues. The discrimination accuracy of ER-positive and ER-negative tissues was 84%.
Programmed death-ligand 1 (PD-L1) expression status is one of the biomarkers for clinical response to cancer immunotherapy [55]. PD-L1 expression status is usually evaluated by visual assessment of immunostained tissue slides and suffers from inter-observer variability. Sha et al. [56] tried to predict PD-L1 status from tissue slides of non-small cell lung cancer and achieved an AUROC of 0.80. The prediction performance was much better for adenocarcinoma than for squamous cell carcinoma.
In addition to ER status, progesterone receptor (PR) and HER2 receptor statuses are also important biomarkers for breast cancer. Rawat et al. [57] tried to predict the statuses of ER, PR, and HER2, which are usually assessed by immunohistochemistry. The AUROCs for ER, PR, and HER2 statuses were 0.88, 0.78, and 0.71, respectively.
Naik et al. [58] enhanced the performance of prediction models for ER, PR, and HER2 statuses in breast cancer by applying the attention mapping method. They obtained the AUROCs of 0.92, 0.81, and 0.778 for ER, PR, and HER2 statuses, respectively.
Spatial transcriptomics technology allowed the expression of multiple genes to be measured from multiple locations within each tissue sample. He et al. [59] trained classifiers based on spatial transcriptomics data and predicted spatial variation in the expression of multiple genes. In 102 of 250 genes, the predictions were positively correlated with the experimentally measured expression. The expression of GNAS, ACTG1, FASN, DDX5, and XBP1 genes was especially well predicted. This study was unique in that the correlation of spatial expression patterns between actual experimental data and DL-based prediction could be examined because the data were obtained through spatial transcriptomics.
Schmauch et al. [60] tried to predict gene expression profiles from multiple tumor types presented by the TCGA program. The study included 8,725 patients with 28 cancer types. An average of 3,627 genes per cancer type was predicted to have altered expression with statistically significant correlation. They also tested the spatial correlation of their prediction results with tissues immunostained for CD3 and CD20. The staining levels were well correlated with the prediction results, confirming the excellent spatial prediction of CD3 and CD20 expression.
Levy-Jurgenson et al. [61] trained DL-based prediction models for the expression level of various RNAs and miRNAs from tissue slides of breast and lung cancers. Five genes for breast cancer and three genes for lung adenocarcinoma were predicted with the AUROCs higher than 0.6. Furthermore, the authors demonstrated that heterogeneity in expression level is a reliable negative predictor of survival. For many cases, highly heterogeneous expression patterns were correlated with poor prognosis.
TREATMENT RESPONSE AND PROGNOSIS
The aforementioned molecular biomarkers offer information for prediction of treatment response and prognosis. Because prediction of these biomarkers was feasible with DL, direct prediction of treatment response and prognosis could be possible. Therefore, many studies have tested DL-based prediction of treatment response and prognosis from H&Estained tissue slides (Table 6).
Hu et al. [62] tried to predict the anti-PD-1 response in melanoma and lung cancer. Because the TCGA dataset did not offer information on immunotherapy responses, they adopted interferon-gamma (INF-╬│) scores as a surrogate for the training of anti-PD-1 response prediction in a DL model. Then, they used their own dataset with anti-PD-1 therapy response data to validate the model. The AUROCs for melanoma and lung cancer were 0.778 and 0.645, respectively. Interestingly, they compared the performance of their INF-╬│-based model with that of a TIL-based model because TIL has been thought as a biomarker for immunotherapy response. The AUROC of the TIL-based model was only 0.58 for melanoma. Therefore, their results indicated that INF-╬│ scores could be a good biomarker to predict anti-PD-1 response.
Johannet et al. [63] directly trained immunotherapy response prediction models on metastasized melanoma tissues of patients treated with anti-CTLA-4, anti-PD-1, or a combination of anti-CTLA-4 and anti-PD-1 therapy. The prediction results were better for lymph nodes than soft tissue, with the AUROCs of 0.857 and 0.583, respectively, and an overall AUROC of 0.691. Furthermore, the prediction results were improved by incorporating clinical data into the model. The AUROC of the combined model was 0.793.
Bychkov et al. [64] trained a model to discriminate low- and high-risk groups of CRC patients directly from H&E-stained tissue microarray images. The DL model yielded an AUROC of 0.69 with a hazard ratio (HR) of 2.3. A visual risk score was assessed by visual examination of tissue slides by three expert pathologists. The AUROC of the visual risk score was only 0.58, with an HR of 1.67. Therefore, the DL model for prognosis prediction outperformed visual assessment by pathologists.
Mobadersany et al. [65] trained a time-to-event model to predict patient outcomes from glioma tissues. Because it was a time-to-event model, HarrellŌĆÖs C index was measured to assess concordance between the model and actual survival rather than AUROC. The model achieved a C index of 0.741. When genomic information including IDH mutation status and 1p/19q co-deletion status were integrated into the model, the C index was increased to 0.781. These results indicate that tissue morphology and genetic alteration status contain complementary information for prediction of survival.
Kather et al. [66] tried to predict the overall survival (OS) of CRC patients. They trained a DL model to classify tissue images into adipose, background, debris, lymphocytes, mucus, smooth muscle, normal colon mucosa, cancer-associated stroma, and tumor tissues. Then, the weighted sum of the activation of the neural network on non-tumor tissue, including adipose tissue, debris, lymphocytes, smooth muscle, and cancer-associated stroma, was calculated as a deep stromal score. The deep stromal score was confirmed to be an independent prognostic factor for OS, with an HR of 1.99. This result demonstrates that non-tumor stromal tissues can provide prognostic information for survival.
Courtiol et al. [67] trained a model to predict the survival time of patients with malignant mesothelioma. The model achieved a C index of 0.643. They also showed that the most relevant image patches for prediction of survival were located in stromal regions rather than tumor regions. The results also indicated that non-tumor stromal tissues are important for prognostic prediction.
Skrede et al. [68] tried to predict the survival of CRC patients with cancer-specific survival as a primary endpoint. They collected huge datasets of four training cohorts and one validation cohort. Ensemble results of 10 total models, five models trained with 10├Ś tissue images and five models trained with 40├Ś tissue images, were used to improve the performance of prediction model for the discrimination of good or poor prognostic groups. The ensemble model could discriminate good or poor prognosis with an HR of 3.84.
Kulkarni et al. [69] proposed a DL method to predict visceral recurrence and disease-specific survival (DSS) from the H&E-stained tissues of primary melanoma. They implemented a complex model that incorporated information from DL-based tissue analysis and morphology-based features. The model predicted distant recurrence with an AUROC of 0.905 and DSS with an HR of 58.7.
From the combined datasets of 10 cancers, including bladder, breast, colon, head and neck, kidney, liver, lung, ovary, and stomach cancers, Wulczyn et al. [70] tried to predict prognosis. They mixed tissue image patches from patients from all 10 cancer groups with good and poor prognoses and then trained a unified model. However, the threshold to determine the low- and high-risk groups of each cancer was set individually for better performance. The overall HR for the 10 cancer types was 1.58. The model could sub-stratify low- and high-risk groups across stage II and III cancers but not stage I and IV cancers. When separately analyzed by cancer type, DSS was predicted for breast, colon, head and neck, kidney, and liver cancers with statistical significance.
Fu et al. [14] also tried prognosis prediction for multiple cancers. They reused features from the Inception v4-based classifier for discrimination of tissue types to train a predictive model for OS. Compared to the prediction based on tumor grade and subtype, the DL model yielded significantly better results for 15 of 18 cancer types, with concordance ranging from 0.53 to 0.67.
Saillard et al. [71] trained predictive models for survival of hepatocellular carcinoma (HCC) patients. Their models could stratify patients with different prognoses even after stratification for other clinicopathologic features, such as disease stage, satellite nodules, alpha-fetoprotein serum level, and vascular invasion. The results indicated that the model could capture unique information from tissue images that is nonredundant with other variables known to affect survival.
Wang et al. [72] adopted a unique approach to extract prognostic information from the dissected lymph nodes of GC patients. By applying a segmentation network, they first extracted lymph node regions and removed other tissues, such as fat and muscle. Then metastasized tumor and normal lymph node tissues were separated with another classifier network. Finally, the area ratio of metastasized tumor vs. lymph node was calculated as a candidate marker for prediction of prognosis. The ratio could discriminate between good and poor prognosis groups with an HR of 2.05.
Wulczyn et al. [73] tried to predict 5-year DSS for patients with stage II and III CRC. They first separated tumor tissue with the Inception-v3-based classifier. Then, tumor tissues were used to train a MobileNet-based prognosis prediction model. The model predicted 5-year DSS with the AUROCs of 0.70 and 0.69 for stage II and III cancer, respectively. When they investigated the correlation of clinicopathologic features and high-risk scores from the DL model, higher T and N categories showed higher risk scores.
Shim et al. [74] tried to predict the recurrence of early-stage lung adenocarcinoma. They adopted a multi-scale approach by combining the feature layers from two independent neural networks analyzing 10├Ś and 40├Ś tissue images. Their model predicted recurrence with the AUROCs of 0.77 and 0.76 for external validation cohorts I and II, respectively. They also found a high probability of recurrence to be associated with tumor necrosis, discohesive tumor cells, and atypical nuclei.
OTHER APPLICATIONS OF DL FOR TISSUE ANALYSIS
Because many pathologic evaluation processes suffer from inter- and intra-observer variability, DL can help to improve the reliability of quantitative evaluation of tissue slides.75 Therefore, in addition to the prediction of molecular biomarkers, DL can be applied to assist many other pathologic tasks. In this section, these other application fields will be briefly introduced with liver tissue assessment as an example. First of all, DL can assist basic diagnosis of HCC by discriminating tumor tissues from normal liver tissues. Pathologists should also discriminate between HCC and cholangiocarcinoma during the assessment of primary liver cancers. A DL-based assistant tool improved the accuracy of the pathologistsŌĆÖ discrimination between HCC and cholangiocarcinoma [76]. Furthermore, tumor tissues can be subclassified by DL depending on differentiation status [12,77]. As these examples showed, DL can provide important tools to assist liver cancer assessment from tissue slides. In addition, liver tissues should be analyzed to evaluate other liver diseases. For the evaluation of nonalcoholic fatty liver disease, histologic changes such as ballooned hepatocytes or fat accumulation can be assessed with DL [78]. DL-based automatic assessment of liver fibrosis can improve liver fibrosis quantification and scoring [79]. During liver transplantation, pathologists should evaluate donor liver biopsies for accepting or discarding the donor liver. DL can help to quantify the percentage of steatosis to evaluate frozen biopsies of donor liver [80]. Therefore, DL can be used to assist almost every aspect of pathologic workflow to improve the objective evaluation of tissue slides.
DISCUSSION
Although these studies clearly demonstrated the potential of DL for prediction of molecular biomarkers, the performance is generally far behind the threshold for clinical applicability. However, we expect that performance will be improved soon because more data for the training of the DL system will be available within a few years. Many studies demonstrated that performance can be enhanced when multi-national and multi-institutional data are collected to be used in training. It has been less than 5 years since digital slide scanners have been widely adopted in hospitals. Therefore, we are still in an early phase of digital pathology, and much more data will likely be accumulated in the near future. It will be interesting to see the improvement in performance with ongoing research.
Small image patch-based classification provides one of the strongest advantages of the DL-based approach. As demonstrated in Figure 2, tumor heterogeneity can be automatically revealed by this approach. When the classification result of each small image patch is overlaid on a WSI, a detailed distribution of tissue regions with different molecular profiles can be easily analyzed. Although spatial sequencing techniques, such as single-cell sequencing and spatial transcriptomics technology, can provide information on tumor heterogeneity [81,82], technical difficulty and high cost limit their application in the clinic. The innate ability of the DL method to reveal spatial tumor heterogeneity can be an alternative tool to these costly methods. Many studies utilized the heterogeneity information provided by DL-based classifiers to investigate the impact of intratumoral heterogeneity [17,20,35,45,61]. Generally, highly heterogeneous tumors impart poorer prognoses than homogenous tumors. Tumor heterogeneity is also a source of difficulty for the correct prediction of molecular biomarkers. When tissue has very heterogeneous classification results for a biomarker, it is difficult to determine whether the tissue is negative or positive for the biomarker. Studies aimed at the correct prediction of biomarkers from highly heterogeneous classification results are necessary to improve the reliability of DL-based molecular biomarker prediction.
The ability of DL to perform detailed analysis of tumor heterogeneity from multiple tissue regions is specifically advantageous for liver cancer. Multiple tumor nodules in the liver can have different grades and heterogenous molecular profiles because they can arise from genetically independent clones [83]. Because tumor grades and molecular profiles are important for prognosis of cancer patients, it is inappropriate to make treatment decisions based on a small portion of heterogeneous tumor tissues. Therefore, detailed analysis of multiple tissue regions in multiple tumor nodules is imperative for accurate assessment of liver cancer. DL can help to quantitatively analyze tumor tissue grades and molecular properties in whole tissue specimens in a cost-effective manner. Improved prognostication and better treatment decision using information from the DL system can enhance treatment response for liver cancer patients.
Another strength of the DL-based approach on the H&E-stained tissue slides is that it can be widely adopted for retrospective analysis of cancer treatment response and prognosis. As described, H&E-stained slides are available for almost all cancer patients and can be used to study the correlation of specific biomarkers with treatment response and prognosis. No additional specimens are necessary for DL-based analysis. Since the cost of DL-based molecular biomarkers prediction is also negligible, retrospective studies will likely be encouraged. Therefore, clinicians could utilize the data from their previous treatment experience with DL models to study the impact of specific molecular biomarkers. Furthermore, the costs of prospective clinical trials can be reduced. Because drug responses significantly differ between patients with different mutational profiles and gene expression profiles, many clinical trials have started to adopt the umbrella platform strategy, which assigns treatment arms based on the specific molecular traits of cancer patients [84,85]. It is very costly to determine the molecular profiles of patients in a trial. If the DL-based approach can be made applicable, the costs of molecular tests for patient assignment will be greatly reduced.
Although the DL approach is very promising, there are some hurdles to clinical adoption. First, the black-box nature of DL limits the interpretability of DL-based prediction results. Because a DNN has millions to billions of numerical parameters to be modified during the training process, it is very hard to understand how a DNN classifies a tissue image patch into a specific class. Without an understanding of the basis of the decision, it will be difficult to trust a DL system. Efforts for explainable artificial intelligence will help to enhance the interpretability of DL systems [86]. Another barrier is the need for an individual DL model for every biomarker for each specific cancer. The generalizability of DL-based prediction models for molecular biomarkers is not very high. Therefore, prediction models for each biomarker should be separately developed for each cancer type. It will take years to develop an entire set of clinically actionable prediction systems for molecular biomarkers in major cancers.
The feasibility of DL-based prediction of molecular cancer biomarkers has been extensively studied thus far. Based on the promising results from these studies, we expect that DL models will be widely adopted to support clinical decisions for management of cancer patients in the near future. A DL system can be applied either as a pre-screening tool or as a post-test quality management tool. With pre-screening, definitive cases will not be further tested to save cost. For quality management, diagnosis and molecular test results can be reassured by DL to enhance the reliability of the pathologic reports. Therefore, DL will be an essential tool in the era of precision oncology.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement1
Supplement1 Print
Print