Hepatorenal syndrome: Current concepts and future perspectives
Article information

Abstract
Hepatorenal syndrome (HRS), a progressive but potentially reversible deterioration of kidney function, remains a major complication in patients with advanced cirrhosis, often leading to death before liver transplantation (LT). Recent updates in the pathophysiology, definition, and classification of HRS have led to a complete revision of the nomenclature and diagnostic criteria for HRS type 1, which was renamed HRS-acute kidney injury (AKI). HRS is characterized by severe impairment of kidney function due to increased splanchnic blood flow, activation of several vasoconstriction factors, severe vasoconstriction of the renal arteries in the absence of kidney histologic abnormalities, nitric oxide dysfunction, and systemic inflammation. Diagnosis of HRS remains a challenge because of the lack of specific diagnostic biomarkers that accurately distinguishes structural from functional AKI, and mainly involves the differential diagnosis from other forms of AKI, particularly acute tubular necrosis. The optimal treatment of HRS is LT. While awaiting LT, treatment options include vasoconstrictor drugs to counteract splanchnic arterial vasodilation and plasma volume expansion by intravenous albumin infusion. In patients with HRS unresponsive to pharmacological treatment and with conventional indications for kidney replacement therapy (KRT), such as volume overload, uremia, or electrolyte imbalances, KRT may be applied as a bridging therapy to transplantation. Other interventions, such as transjugular intrahepatic portosystemic shunt, and artificial liver support systems have a very limited role in improving outcomes in HRS. Although recently developed novel therapies have potential to improve outcomes of patients with HRS, further studies are warranted to validate the efficacy of these novel agents.
INTRODUCTION
Hepatorenal syndrome (HRS) is a serious complication of end-stage cirrhosis and portal hypertension that is characterized by increased splanchnic blood flow, a state of decreased central volume, kidney blood flow and glomerular filtration rate (GFR) [1]. Hepatorenal syndrome is typically diagnosed when there is a marked reduction in GFR, and in the absence of evidence of intrinsic kidney diseases, such as hematuria, proteinuria, or abnormal kidney ultrasonography. This is contrary to what occurs in most cases of intrinsic kidney damage, in which there are marked changes in kidney histology [1,2]. This review focuses on several conceptual issues that have emerged in the hepatorenal field.
DEFINITION OF HEPATORENAL SYNDROME
The definition of HRS has significantly evolved over the past several decades (Table 1). In 1996, the International Club of Ascites (ICA) defined acute kidney injury (AKI) in cirrhosis as an increase in serum creatinine of ≥50% from baseline to ≥1.5 mg/dL [3]. Other important components of AKI in cirrhosis included oliguria, as well as proteinuria <500 mg/dL. In 2007, HRS was further classified into two types: type 1, characterized by a rapid deterioration of kidney function by doubling of initial serum creatinine to ≥2.5 mg/dL or a 50% reduction in less than 2 weeks in the initial 24-hour creatinine clearance to below 20 mL/min that often occurs due to a precipitating event; and type 2, in which kidney failure progression did not meet the criteria for type 1. Importantly, urinary sodium and oliguria were removed from the new diagnostic criteria [4]. Several studies indicating that the diagnosis of AKI in patients with cirrhosis, based on an absolute increase in serum creatinine by ≥0.3 mg/dL or 50% from baseline, leads to earlier identification of patients with poorer outcomes led to the ICA to revise the definition of HRS in 2015, incorporating a new definition and classification of AKI with modifications (Table 2) [5-12]. Serum creatinine obtained in the previous 3 months can be used as baseline when a baseline level obtained during the previous 7 days is not available. Although oliguria was not included in the definition of AKI in patients with cirrhosis, a study indicating that urine output was found to be significantly associated with adverse outcomes in patients with AKI and cirrhosis led to calls for a new definition and overall a new classification for HRS that expands on the 2015 ICA consensus document [13,14]. Most recently, the ICA completely revised the nomenclature and diagnostic criteria for HRS type 1, which is now called HRS-AKI [12]. Results of several studies showed that the higher the initial serum creatinine level at the start of treatment, the lower the probability of HRS reversal [15,16]. This led to the ICA removing the minimum creatinine value for diagnosis, and therefore HRS-AKI can be diagnosed even when the serum creatinine level is below 2.5 mg/dL. Functional kidney injury that does not meet the criteria of HRS-AKI is termed HRS-NAKI (i.e., non-AKI), of which NAKI is further divided into HRS-acute kidney disease (HRS-AKD) if the estimated glomerular filtration rate (eGFR) is below 60 mL/min/1.73m2 for less than 3 months and HRS-chronic kidney disease (HRS-CKD) if eGFR is below 60 mL/min/1.73m2 for more than 3 months [14].

Previous and current definitions of hepatorenal syndrome

PATHOPHYSIOLOGY
The pathophysiology of HRS is characterized by reduced systemic vascular resistance due to splanchnic arterial vasodilation, which occurs secondary to portal hypertension, a key feature of advanced cirrhosis. However, recent studies have suggested that a systemic inflammatory state may lead to an increase in the release of inflammatory mediators, and therefore may play a role in the circulatory and kidney dysfunction observed in HRS [17,18]. Therefore, it is now recognized that HRS not only involves circulatory dysfunction but also systemic inflammation (Fig. 1).

Pathophysiology of hepatorenal syndrome. PAMPs, pathogen-associated molecular patterns; DAMPs, damage-associated molecular patterns; RAAS, renin-angiotensin-aldosterone system; SNS, sympathetic nervous system; AVP, arginine vasopressin.
Circulatory dysfunction
End-stage liver disease resulting in cirrhosis leads to increased intrahepatic vascular resistance, which subsequently causes splanchnic vasodilation triggered by increased production of vasodilators including nitric oxide, prostacyclins, carbon monoxide, and endocannabinoids. Splanchnic vasodilation subsequently leads to decreased vascular resistance and reduced effective arterial blood volume (EABV). Although the heart is able to compensate for this decrease in EABV in the early stages of cirrhosis by increasing cardiac output, but subsequent development of cirrhotic cardiomyopathy, aggravation of portal hypertension and splanchnic vasodilation results in effective arterial hypovolemia and arterial hypotension [19]. This decrease in EABV subsequently activates various vasoconstriction factors that include the renin-angiotensin-aldosterone system (RAAS) [20], the sympathetic nervous system (SNS) [21], and the non-osmotic secretion of arginine vasopressin. Although these vasoconstriction factors assist in maintaining arterial pressure near normal limits, their activation has detrimental effects on kidney function, resulting in renal vasoconstriction, impaired solute-free water excretion, and subsequent decline in kidney function. The kidneys are also able to compensate for such changes during earlier stages, owing to the vasodilatory effects of renal prostaglandins (prostaglandins E2 and I2) on afferent renal arterioles. This maintains glomerular pressure despite reduced renal blood flow (RBF). Progression of liver disease and the use of concomitant non-steroidal anti-inflammatory drugs that inhibit prostaglandin synthesis disrupts this balance and, therefore, causes AKI [22].
Diastolic dysfunction may be present in up to 60% of patients with cirrhosis; however, the relationship between diastolic and circulatory dysfunction or development of HRS has not been demonstrated [23]. Nevertheless, decreased cardiac output in patients with cirrhotic cardiomyopathy is associated with the development of kidney hypoperfusion and HRS. For example, in a study of 66 patients who had cirrhosis with ascites and normal serum creatinine levels, baseline mean arterial pressure and cardiac output were significantly higher in patients who did not develop HRS than in those who developed HRS. Plasma renin activity and cardiac output were independent predictors of HRS. Complications occurred in the setting of a significant reduction in mean arterial pressure, cardiac output, and wedged pulmonary pressure, as well as an increase in plasma renin activity, norepinephrine concentration, and hepatic venous pressure gradient. In another study of 23 patients with spontaneous bacterial peritonitis (SBP) at diagnosis and after resolution of infection, those who developed HRS had a significantly lower cardiac output at the time of diagnosis of SBP, compared with those who did not develop HRS, indicating a relationship between HRS and diminished cardiac output [24].
The pathophysiological hallmark of HRS is vasoconstriction of the renal circulation [27-30]. Marked renal vasoconstriction in patients with HRS has been demonstrated in a number of studies [27-30]. This phenomenon may most likely be due to several factors and may involve alterations in systemic hemodynamics, activation of multiple vasoconstrictor factors, and suppression of vasodilatory factors that act on renal circulation. Two major vasoconstrictor systems that are important in this pathophysiological process is the RAAS and the SNS [20,21]. In several studies of patients with cirrhosis, activity of the RAAS, as estimated by plasma renin activity, was shown to increase from compensated to decompensated cirrhosis. Peak activity was seen in patients with HRS and it was shown to correlate inversely with kidney function [19,20,22]. Moreover, in patients with infection associated HRS, patients with higher RAAS activity had a significantly lower probability of HRS reversal than those with lower RAAS activity [31]. Plasma levels of norepinephrine, which reflects SNS activity, are increased in patients with HRS than in those with ascites and intact kidney function, and were shown to be inversely correlated with GFR [21]. However, considering that both RAAS and SNS are two vasoconstrictor systems that act to increase arterial blood pressure and counteract splanchnic vasodilation, studies have been unable to assess whether the blockade of these RAAS and SNS lead to improved outcomes in patients with cirrhosis [32]. Other than the aforementioned vasoconstrictor systems, other factors with a potential role in kidney vasoconstriction in HRS include endothelin, cysteinyl leukotrienes, and prostaglandins [33-35].
Nitric oxide dysfunction
In cirrhosis, a reduction in RBF is also partly due to either excessive or insufficient nitric oxide (NO) production [36-39]. Excess NO production results in splanchnic vasodilation, reduced EABV, RAAS, and SNS activation, and renal vasoconstriction. However, insufficient NO release may also cause reduced RBF. This may be partly due to the increased production of dimethylarginines, such as symmetric (SDMA) and asymmetric dimethylarginine (ADMA). ADMA levels are increased in advanced liver disease, and therefore NO synthesis from NO synthase (NOS) is inhibited, and therefore RBF is compromised [36-38,40]. SDMA, an ADMA isomer, also increases in the setting of decreased hepatic and kidney function. High concentrations of SDMA also reduces NO production, resulting in reduced RBF. This has led to some studies indicating that SDMA may be a potential marker for HRS [36]. Indeed, several studies have indicated that both SDMA and ADMA are potential independent predictors of measured GFR in cirrhotic patients [41].
Systemic inflammation
It is now recognized that systemic inflammation also plays a part in HRS pathophysiology. Systemic inflammatory response syndrome has been observed in almost half of patients with HRS-AKI, independent of the presence of actual infection [42]. In particular, those with the most extensive baseline systemic inflammation also had the highest risk of liver failure development and mortality [43]. Plasma levels of pro-inflammatory cytokines (interleukin-6 [IL-6] and tumor necrosis factor-α [TNF-α]), and urinary levels of monocyte chemoattractant protein-1 are increased in patients with HRS-AKI than in those with decompensated cirrhosis without AKI and those with AKI secondary to pre-renal azotemia [44].
The main mechanism by which the systemic inflammatory state primarily contributes to the pathogenesis of HRS is the translocation of gut bacteria from the gut to mesenteric lymph nodes due to altered intestinal permeability [45]. This bacterial translocation not only induces increased levels of pro-inflammatory cytokines [45], in particular IL-6 and TNF-a [46,47], but also increased levels of various vasodilating factors, such as NO [48], which contribute to the decreased EABV, as well as a wide spectrum of molecules (pathogen-associated molecular patterns [PAMPs] and damage-associated molecular patterns [DAMPs]) that are responsible for inducing inflammatory responses through activation of pattern recognition receptors such as toll-like receptors (TLRs). PAMPs are products of bacteria that include lipopolysaccharide, flagellin, and nigericin, whereas DAMPS are intracellular components released from injured hepatocytes that include high-mobility group protein B1, heat shock proteins, and hyaluronic acid. Not only are both PAMPs and DAMPs known to have systemic effects by promoting inflammation and the release of proinflammatory cytokines, but both molecules may also have direct effects on the kidney. For example, in a study of patients with kidney dysfunction and cirrhosis, patients showed increased renal expression and urinary excretion of TLR4, suggesting a potential role of TLR4 as a mediator of kidney injury [49]. Moreover, gut decontamination in rodent models of cirrhosis has been shown to reduce renal TLR4 expression and subsequently prevent kidney dysfunction, suggesting that exposure to PAMPs from gut bacterial translocation may increase TLR4 expression in the kidneys [50].
DIFFERENTIAL DIAGNOSIS AND BIOMARKERS
As the treatment of AKI in patients with cirrhosis depends on the type of AKI, determining the etiology is essential [25,51]. Although the differential diagnosis of AKI in patients with cirrhosis is broadly similar to that in other patient populations, the differential diagnosis is often not so straightforward. In addition to HRS, other types of AKI that can occur include volume-responsive pre-renal AKI due to infection, hypovolemia, vasodilators, obstructive post-renal AKI, and intra-renal AKI that may be caused by toxin or ischemia induced acute tubular necrosis (ATN), or glomerulonephritis. Considering that patients with ATN and HRS have the worst survival among those with AKI and cirrhosis [52], accurate differential diagnosis of the etiology is important.
HRS remains a diagnosis of exclusion. A key component in HRS diagnosis is exclusion of structural kidney damage, which relies on urine microscopy and urine sodium excretion. Other requirements include the absence of shock, proteinuria (>500 mg/day), and microscopic hematuria (>50 red blood cells per high power field), along with normal kidney morphology on ultrasonography. However, possibly due to systemic inflammation that can also cause ATN, differentiating between ATN and HRS is often very difficult. Although urinary sodium (>40 mEq/L), fractional excretion of sodium (FeNa >2%), and low urine osmolality (<400 mOsm/L) are suggestive of ATN, other conditions, such as the use of diuretics that are commonly used in patients with large volume ascites, may confound the interpretation of FeNa [53]. Moreover, low FeNa was also found in biopsy proven-ATN [54], and therefore urinary sodium and FeNa are no longer part of the most recent diagnostic criteria of HRS-AKI [14]. A more useful marker in differentiating between ATN and HRS may be the fractional excretion of urea [55,56], because unlike sodium, reabsorption of urea occurs primarily in the proximal tubules of the kidney, and therefore is not affected by commonly used diuretics such as loop diuretics and spironolactone, which act in the loop of Henle and distal convoluted tubules, respectively.
Several novel biomarkers that may be useful in the differential diagnosis of AKI in patients with cirrhosis have recently been investigated [57-62]. Most of these biomarkers originate from kidney tubular proteins released during cell damage, upregulated during kidney injury, proteins with diminished tubular reabsorption, and markers of inflammation. Of the above markers, to date, the most widely investigated is urinary neutrophil gelatinase associated lipocalin (NGAL) [58-62]. In a multicenter, prospective cohort study involving 188 patients with AKI and cirrhosis, median values of urinary NGAL, IL-18, kidney injury molecule-1, liver-type fatty acid binding protein, and albumin were all elevated in patients with ATN [60]. In another study involving 241 patients with cirrhosis, urinary NGAL levels were markedly higher in patients with ATN than in those with pre-renal azotemia, CKD, and HRS [59]. In a more recent study involving 320 patients with AKI hospitalized for decompensated cirrhosis, urinary NGAL measured at day 3 had the greatest accuracy for differential diagnosis between ATN and other etiologies of AKI [61].
Not only are biomarkers important for the differential diagnosis of AKI, but they may also play an important role in predicting treatment response of HRS, and even for prognosis [63,64]. For example, in 162 patients with AKI and cirrhosis, not only was urinary NGAL an adequate biomarker in the differential diagnosis of AKI, but it also predicted the response to terlipressin and albumin in patients with HRS-AKI, and was also an independent predictor of in-hospital mortality [63]. Similarly, in a study consisting of 213 United States (US) hospitalized patients with decompensated cirrhosis, not only did urinary NGAL differentiate the type of AKI in cirrhosis, but also significantly predicted 90-day transplant-free survival, and outperformed Model for End-Stage Liver Disease score in terms of survival prediction [64].
Although the most ideal biomarker would be one that distinguishes structural from functional AKI, but in reality, no biomarkers to date perform optimally in the differential diagnosis of AKI in patients with cirrhosis. Further validation studies are warranted for their generalized applications.
RISK FACTORS AND PREVENTION
The most common risk factors for HRS are those related to systemic inflammation and acute hemodynamic changes. Therefore, the most commonly known precipitants of HRS are SBP, other systemic infections, and large volume paracentesis without albumin administration. HRS develops in as many as 30% of patients with SBP, and is associated with significantly worse outcomes [31,65]. Infection-associated HRS may be prevented by administration of intravenous (IV) albumin in addition to antibiotic treatment in the setting of SBP and may also reduce overall mortality [66,67]. In the setting of SBP, IV albumin may be administered 1.5 g/kg on day 1 followed by 1 g/kg on day 3. In patients undergoing large-volume paracentesis (>5 L), albumin administration has been shown to decrease the incidence of HRS [68]. However, data on whether albumin prevents HRS or improves overall survival has been conflicting [69-73]. For example, in the ANSWER (human Albumin for the treatmeNt of aScites in patients With hEpatic ciRrhosis) study, long-term administration of human albumin was associated with improved overall 18-month survival compared to standard medical treatment [72]. However, in the MACHT (midodrine and albumin for cirrhotic patients in the waiting list for liver transplantation) study, treatment with midodrine and albumin failed to prevent complications of cirrhosis or improve survival [73]. Most recently, in the ATTIRE (Albumin to Prevent Infection in Chronic Liver Failure) trial that investigated whether higher doses of albumin therapy to increase and maintain serum albumin levels to 30 g/L or more improved outcomes in hospitalized patients with cirrhosis, the results were largely disappointing, and therefore, supporting the need for a re-evaluation of the use of albumin in patients with cirrhosis [74]. The ongoing PRECIOSA12 (Effects of Long term Administration of Human Albumin in Subjects With Decompensated Cirrhosis and Ascites) trial will hopefully clarify the role of long term albumin use in this population [75].
β-blockers are effective in preventing variceal bleeding, which can precipitate HRS. Although they are widely used in patients with cirrhosis and portal hypertension [76], therapy must be individualized based on the severity of hepatic decompensation [77]. In patients with compensated cirrhosis, treatment with β-blockers was associated with a preservation in kidney function, and an increase in decompensationfree survival, mainly by reducing the incidence of ascites [78]. However, in patients with decompensated cirrhosis with ascites, reports have been conflicting. While a recent meta-analysis suggested that the use of β-blockers in patients with cirrhosis and ascites was not associated with a significant increase in mortality [79], some reports have suggested that the decrease in cardiac output caused by β-blockers could precipitate AKI, and therefore increase mortality in this patient group [77,80]. Therefore, clinicians should carefully weigh the risks and benefits of continuation of β-blockers in patients with cirrhosis.
In patients with ascites and a high risk of developing SBP, as determined by a low ascitic fluid protein (<1.5 g/dL), concomitant advanced liver failure (Child-Pugh score ≥9 points with serum bilirubin level ≥3 mg/dL) or kidney dysfunction (serum creatinine level ≥1.2 mg/dL, blood urea nitrogen level ≥25 mg/dL, or serum sodium level ≤130 mEq/L), antibiotic prophylaxis using either norfloxacin or rifaximin has shown to prevent the development of SBP and HRS, as well as reduce overall mortality [68,81].
TREATMENT OF HEPATORENAL SYNDROME
The management of AKI in patients with cirrhosis should begin immediately once a diagnosis has been made and the etiology of AKI identified because patients with AKI and cirrhosis often deteriorate rapidly (Fig. 2). Once a diagnosis of AKI has been made, management of AKI should typically begin with a fluid challenge of 20–25% IV albumin at 1 g/kg/day for 2 days and withdrawal of any diuretics (expert opinion, not evidence-based) [12]. Low volume therapeutic paracentesis with albumin to control ascites should also be performed if necessary [82,83]. This not only rules out pre-renal azotemia, but also promotes early circulating volume expansion in the context of reduced EABV. The initial phase of treatment also consists of temporary discontinuation of non-selective β-blockers given their negative inotropic effect [84,85], and other potential nephrotoxic agents and vasodilators.
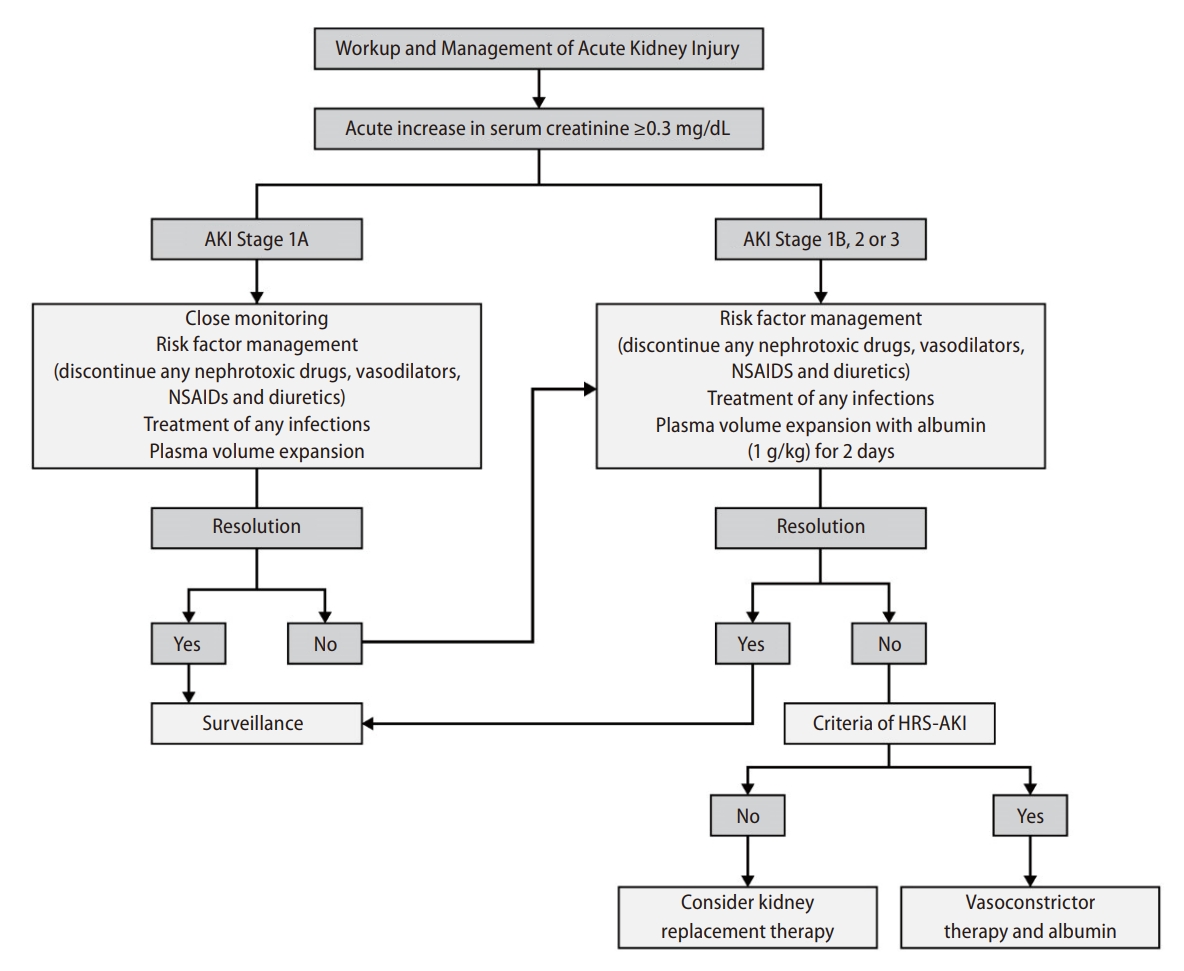
Algorithm for the management of acute kidney injury in patients with cirrhosis. AKI stages 1A and 1B are adaptations of the International Club of Ascites definitions of AKI stages by the European Association for the Study of the Liver [101]. NSAIDs, non-steroidal anti-inflammatory drugs; AKI, acute kidney injury; HRS-AKI, hepatorenal syndrome-acute kidney injury.
Pharmacologic therapy Vasoconstrictors
The rationale behind the use of vasoconstrictors in the treatment of HRS is to counteract splanchnic arterial vasodilation [25,86]. Of the currently available vasoconstrictors, terlipressin, a synthetic vasopressin analog with a predominant vasopressin 1A receptor effect acting primarily as a splanchnic vasoconstrictor [87], is the most commonly used vasopressin analog.
To date, terlipressin is the vasopressin with the most convincing data to date [88]. Several positive results from clinical trials have led to the US Food and Drug Administration recently approving the use of terlipressin in the US for improving kidney function in patients with HRS. Terlipressin is generally administered by IV boluses at starting doses of 0.5–1 mg every 4–6 hours. The dose can be increased to a maximum of 2 mg every 4 hours in cases of nonresponse, defined as less than a 25% reduction in serum creatinine level after 3 days and no side effects occur [89-91]. Doses should be maintained for a maximum of 14 days depending on responses to treatment. Doses of terlipressin in combination with albumin should be continued until serum creatinine reaches a final value <1.5 mg/dL, or until baseline creatinine level. If patients show nonresponse or partial response, the treatment should be discontinued within 14 days. The efficacy of continuous infusion of terlipressin has been supported in a single center study of 78 patients, where continuous IV terlipressin infusion was shown to be not only better tolerated than IV boluses, but was also effective at doses lower than those required for IV bolus administration [92]. The INFUSE (Terlipressin for HRS-AKI in Liver Transplant Candidates) trial that will evaluate the use of continuous terlipressin infusion in patients on the liver transplant waiting list with HRS-AKI is currently ongoing [93].
According to two previous randomized controlled clinical trials performed in Europe, the response rate of terlipressin plus albumin is around 50% [92,94]. Most recently, the efficacy and safety of terlipressin plus albumin for the treatment of type 1 HRS have been proven in a multicenter phase 3 trial, in which enrolled patients were randomly assigned in a 2:1 ratio to receive terlipressin or placebo for up to 14 days. In this trial involving 300 patients with cirrhosis and type 1 HRS, terlipressin was more effective than placebo in improving kidney function [95]. Despite the high response rates of terlipressin plus albumin, recurrence of HRS is not uncommon, with recurrences occurring in <20% of patients with type 1 HRS. These patients may be re-treated with vasoconstrictors and albumin. The patient’s response to terlipressin is not only important for HRS reversal, but it has also shown to be an important prognostic factor in liver transplantation (LT) patients [96]. For example, in two cohorts of patients with cirrhosis listed for LT, one with and one without HRS-AKI, response to terlipressin and albumin reduced the need for KRT after LT, and also reduced the risk of CKD at 1 year after LT [98]. Factors associated with lower response to terlipressin and albumin include higher baseline serum creatinine, urinary NGAL and serum bilirubin, lower increases in arterial pressure, presence of systemic inflammatory response syndrome, and more severe acute on chronic liver failure grade [15,63,97-99].
Common side effects of terlipressin include diarrhea and abdominal pain, which are reported in around 10–20% of patients. More serious side effects are related to vasoconstriction with a risk of myocardial infarction and intestinal ischemia, with a rate of 2–13% [100]. In the recent CONFIRM trial, the use of terlipressin was also associated with a higher risk of respiratory failure. Patients on terlipressin should be monitored for signs of ischemia while on therapy, and the drug should be avoided in patients with a history of coronary artery disease or peripheral artery disease [101]. Furthermore, as the response to treatment is attenuated in patients with higher degrees of kidney injury and acute on chronic liver failure grade [99], the risk-benefit of administering vasoconstrictors in combination with IV albumin should be carefully considered.
Other vasoconstrictor treatment options include norepinephrine, and the combination of midodrine and octreotide. Norepinephrine is a systemic vasoconstrictor that acts through the activation of α-1 adrenergic receptors on vascular smooth muscle cells. Norepinephrine is administered at 0.5–3 mg/h continuous IV infusion, titrating dosing to achieve an increase of 10 mmHg in mean arterial pressure [89-91,102,103]. Norepinephrine in combination with albumin is also effective and safe [104], with response rates ranging from 39–70% [105-107]. It is a cheaper drug than terlipressin; however, unlike terlipressin, which can be administered peripherally, norepinephrine can only be administered through a central venous line. When using norepinephrine, close monitoring for tachyarrhythmias or bradycardia is needed [94].
A combination of midodrine, an α-adrenergic agonist, plus octreotide, a somatostatin analogue, may also be used. Midodrine is administered as 7.5 mg up to 12.5 mg orally three times a day; doses should be titrated to achieve an increase of 15 mmHg in mean arterial pressure [89-91,102,103]. Octreotide is administered as 100–200 μg subcutaneously every 8 hours. In case of nonresponse, doses of both drugs can be increased on day 3 of treatment. In a pilot study, the combination of midodrine and octreotide, plus albumin restored kidney function in approximately 40% of patients with HRS [108].
Several meta-analyses have evaluated and compared the efficacy of vasoconstrictors, where studies have shown that terlipressin, in combination with IV albumin, has the highest efficacy [16,94,95,105-107,109-111]. Although comparisons of terlipressin with norepinephrine and norepinephrine with octreotide and midodrine did not show any significant differences, terlipressin had better efficacy in reversing HRS than midodrine plus octreotide. In terms of overall mortality, meta-analyses results have revealed that most vasoconstrictors did not show any significant reduction in overall mortality [100,112]. Although these results are disappointing, it must be noted that interventions to improve kidney function do not improve the underlying poor hepatic function in patients with HRS.
Albumin
Albumin is often administered in combination with vasoconstrictors to counteract the reduction in EABV and improve cardiac contractility [113-115]. The efficacy of terlipressin when administered in combination with albumin has been proven in a large number of studies. In the only study in which terlipressin was used alone for the treatment of HRS, the efficacy of terlipressin was much lower than when it was used in combination with albumin [116]. This may be due to the ability of albumin to maintain or increase cardiac output even in the most advanced phases of liver disease [114]. The recommended dose is generally 20–40 g IV once daily after the initial dose of albumin is administered as 1 g/kg/day for 2 days [89-91,102,103].
Accumulating experimental and clinical evidence is suggesting that not only is albumin capable of increasing systemic vascular resistance and cardiac output, its capacity of exerting anti-oxidant and anti-inflammatory actions also plays a role in mitigating the inflammatory state associated with HRS [113,115,117]. Albumin is able to bind a wide range of substances, including various bacterial products, bile acids, cytokines, nitric oxide, and endotoxins [118]. Although this results in a significant reduction in serum creatinine levels in patients with HRS [119], in patients refractory to vasoconstrictors, improvements in kidney function and systemic hemodynamics are not observed upon administration of IV albumin, despite a reduction in NO concentrations [120].
Role of kidney replacement therapy in the treatment of hepatorenal syndrome
Although there is no definite role of KRT in the treatment of AKI in patients with cirrhosis, KRT may be indicated in those unresponsive to pharmacological treatment and with conventional indications for KRT such as volume overload, uremia, or electrolyte imbalances [121], as well as a bridging therapy to transplantation [122]. According to a retrospective study that involved HRS patients who were non-responders to vasoconstrictor therapy, KRT did not provide any significant improvements in either 30-day or 180-day survival, and only led to significantly longer hospital stays [123]. The ideal timing and the best modality of KRT has not been studied in patients with cirrhosis, and so the decision to initiate KRT should be made on clinical grounds, such as worsening kidney function with intractable volume overload, diuretic intolerance or resistance, or medically refractory electrolyte disturbances. To prevent fluid accumulation, KRT should also be considered if the daily fluid balance cannot be maintained, regardless of urine output. Continuous kidney replacement therapy (CKRT) should be used in hemodynamically unstable patients, and also has the advantage of not increase intracranial pressure, which is in contrast to conventional KRT. In cirrhotic patients with hyperammonemia and encephalopathy, CKRT may be used to mitigate cerebral edema and encephalopathy, but the cut-off ammonia level requiring initiation of CKRT is unknown [124].
In the setting of CKRT, the molecular adsorbent recirculating system (MARS) is a potential therapeutic modality. MARS is an extracorporeal liver support system based on albumin dialysis, given that albumin is one of the most important molecules involved in the detoxification and the liver regulation process. MARS removes albumin-bound toxins which may have detrimental effects on hepatocytes and other organs, as well as other water-soluble cytokines [125]. A small prospective controlled trial involving 13 patients with cirrhosis and HRS demonstrated that mortality at day 7 was significantly lower than in the control group [119], suggesting that the removal of albumin-bound substances with the MARS method may contribute to the treatment of HRS. However, in a larger study involving 189 patients with acute-on-chronic liver failure, although MARS significantly decreased serum creatinine levels at day 4 compared to standard medical treatment, there was no significant difference in the 28-day mortality between the two treatment groups [126]. Given the conflicting results, further observational and prospective controlled trials are needed for the generalized application of supportive detoxification therapies.
Transjugular intrahepatic portosystemic shunt
In theory, a transjugular intrahepatic portosystemic shunt (TIPS) that connects the portal vein with one of the hepatic veins may improve kidney function in HRS by decreasing portal hypertension and reducing and reversing the hemodynamic changes that precipitate HRS. However, there is only a paucity of studies that have looked into the role of TIPS in HRS, so its use remains controversial in this population group. Although results of a few studies have demonstrated that TIPS could improve kidney function by improving serum creatinine, serum sodium, and urine output, the relative liver ischemia that immediately follows TIPS insertion could potentially precipitate hepatic failure in patients with predisposed severe liver dysfunction [127]. Hopefully, the ongoing Liver-HERO (HRS-AKI Treatment With Tips in Patients with Cirrhosis) trial that compares the effectiveness and safety of TIPS implantation in patients with stage 2 and 3 HRS-AKI and liver cirrhosis with standard therapy of terlipressin and albumin will clarify the role of TIPS for use in this patient population [128].
Liver transplantation
LT remains the definitive treatment for HRS. Although simultaneous liver-kidney transplantation (SKLT) is the procedure of choice if native kidney function recovery is not expected after LT, the decision to perform SKLT versus LT remains a challenge. Predicting the recovery of impaired kidney function is challenging because various factors, particularly the duration of kidney injury, contributes to kidney prognosis [129]. In the US, the Organ Procurement and Transplantation Network policy for simultaneous liver-kidney organ allocation requires an eGFR of ≤25 mL/min/1.73m2 for 6 weeks or a period of kidney replacement therapy of ≥6 weeks in patients with AKI, presence of CKD G3b, which is defined as eGFR of <44 mL/min/1.73m2 for >90 days, or comorbid presence of metabolic diseases [130]. European guidelines recommend that patients with end-stage liver disease who also have CKD G4 or 5, defined as eGFR <30 mL/min/1.73m2, or type 1 HRS requiring kidney replacement therapy of >8–12 weeks and patients with kidney biopsy samples revealing >30% glomerulosclerosis and fibrosis should receive SLKT [90]. Nevertheless, in approximately 10% of patients who receive LT may have persistent or progressive kidney dysfunction even after a successful transplant [131]. In particular, patients with ATN are at higher risk of CKD post-transplant, and the lack of ideal biomarkers often results in misdiagnosis [132].
CONCLUSIONS
There have been considerable improvements in the diagnosis and management of HRS, as well as evolving definitions, advances in pathophysiological understanding, and biomarker discovery. Lowering of the serum creatinine level threshold for the diagnosis of HRS-AKI has allowed for earlier recognition and treatment. In terms of pathophysiology, it is now recognized that HRS not only involves circulatory dysfunction, but also systemic inflammation. New insights into the pathophysiological basis have allowed for further investigation into novel therapeutic agents that target specific pathophysiological pathways. Although the differential diagnosis of AKI in patients with cirrhosis is difficult, recent studies of novel biomarkers have allowed for further investigations into tools that may assist the clinician in the diagnosis and management of HRS-AKI. Moreover, the findings from recent large-scale randomized clinical trials have further supported the use of terlipressin. While LT remains the optimal treatment option, particularly in patients with a high risk of persistent kidney dysfunction, SLKT may be warranted over LT alone. During the patient’s time on the liver transplant waiting list, CKRT may be considered as a bridging therapy to transplantation.
Liver transplantation
Despite the recent advances in HRS, much remains to be uncovered. Although there have been consistent efforts into updating the definition of HRS over the past decades, the clinical implications of the newly proposed diagnostic criteria are still unclear. Hopefully, results from future validation studies will further refine the diagnostic criteria to allow for earlier recognition and thus, management of HRS. Although it is now recognized that systemic inflammation plays an important role in the pathophysiology of HRS, the exact mechanisms by which systemic inflammation leads to HRS remain to be elucidated. While novel biomarkers that differentiates structural from functional AKI have been recently investigated, their predictive performances are far from optimal, and therefore further validation studies are needed. Not only do these novel biomarkers have the potential to differentiate AKI etiology, but they also have the potential to predict treatment response of HRS, as well as predict the prognosis of patient with HRS. Despite the positive results from several randomized clinical trials that have supported the use of terlipressin in patients with HRS-AKI, terlipressin is a drug not without significant side effects, and therefore the risk-benefit of administering terlipressin in combination with IV albumin should be carefully considered. Investigation into novel biomarkers that will not only allow for adequate selection of patients for vasoconstrictor therapy and proportional albumin use to reduce risk of adverse events, but also assist in predicting the reversibility of kidney dysfunction after LT is warranted.
Notes
Authors’ contribution
Conception and design: J.W. Chang; Writing, review, and/or revision of the manuscript: C.Y. Jung and J.W. Chang; Administrative, technical, or material support: J.W. Chang; Study supervision: J.W. Chang.
Conflicts of Interest
The authors have no conflicts to disclose.
Acknowledgements
The authors thank the Medical Illustration & Design team of the Medical Research Support Services of Yonsei University College of Medicine for all artistic support related to this work.
Abbreviations
ADMA
asymmetric dimethylarginine
AKD
acute kidney disease
AKI
acute kidney injury
ATN
acute tubular necrosis
CKD
chronic kidney disease
CKRT
continuous kidney replacement therapy
DAMP
damage-associated molecular pattern
EABV
effective arterial blood volume
eGFR
estimated glomerular filtration
FeNa
fractional excretion of sodium
GFR
glomerular filtration rate
HRS
hepatorenal syndrome
ICA
International Club of Ascites
IL-6
interleukin-6
IV
intravenous
KRT
kidney replacement therapy
LT
liver transplantation
MARS
molecular adsorbent recirculating system
NAKI
non-AKI
NGAL
neutrophil gelatinase associated lipocalin
NO
nitric oxide
NOS
nitric oxide synthase
PAMP
pathogen-associated molecular pattern
RAAS
renin-angiotensin-aldosterone system
RBF
renal blood flow
SBP
spontaneous bacterial peritonitis
SDMA
symmetric dimethylarginine
SKLT
simultaneous liver-kidney transplantation
SNS
sympathetic nervous system
TIPS
transjugular intrahepatic portosystemic shunt
TLR
tool-like receptor
TNF-a
tumor necrosis factor-α