Correspondence on Editorial regarding “Identification of signature gene set as highly accurate determination of MASLD progression”
Article information

Dear Editor,
We express our deep gratitude to Dr. Pirola and Dr. Sookoian for their interest in our research [1,2]. The severity of metabolic-associated steatohepatitis liver disease (MASLD), which is characterized by the accumulation of fat in the liver, inflammation, and liver cell damage, is significant. If left untreated, MASLD can progress to more serious conditions, such as cirrhosis and liver cancer [3]. This underscores the importance of disease detection and identifying pharmacological candidates with therapeutic potential. Various non-invasive biomarkers, obtained from serum and identified through imaging techniques, are employed to diagnose diseases and ascertain disease progression [4]. However, in pursuit of more sensitive and precise diagnostics, omics-based biomarkers are rapidly gaining prominence in conjunction with these assessments [5].
Omics-based biomarkers for MASLD represent a comprehensive approach to understanding and diagnosing complex disease conditions by examining a vast array of biological molecules that reflect the state of a cell, tissue, or organism. In our previous study involving tissue samples from 134 patients with MASLD, we identified a set of genes (CAPG, HYAL3, WIPI1, TREM2, SPP1, and RNASE6) that could collectively serve as a diagnostic panel to accurately distinguish MASLD progression through multi-omics analysis, including genomics, epigenomics, and transcriptomics.
We validated the effectiveness of the signature gene set in accurately differentiating discrete cohorts, revealing its substantial discriminatory capability. This gene set enables the stratification of subjects into severity-based subgroups within the examined populations. The ability to distinguish disease severity using signature genes is underpinned by the progressive elevation in their expression profiles, which correlates with the advancement of the disease state. Therefore, we propose that the signature gene set is better suited for assessing the overall severity of MASLD-associated diseases than for identifying a specific stage of MASLD.
Examining the applicability of a signature gene set identified by analyzing omics data obtained from patient tissues for non-invasive assessments is essential for transforming it into viable clinical biomarkers. We investigated whether the signature genes could also be applied to liquid biopsy samples from MASLD-associated disease by examining scRNAseq data from blood samples of patients with MASLD (Fig. 1A) [6]. In our previous study, we had already established that the expression of signature genes was elevated in cell-free RNAs of blood from patients with MASLD compared to that in healthy individuals. Consistent with this, we found that, except for SPP1, the signature genes were also detectable in scRNA data from blood samples of patients with MASLD. Notably, they were predominantly found in the dendritic cells, granulocyte-monocyte progenitors, monocytes, and pre-B cells. Further, we observed that signature gene expression was discernible in blood samples from both patients with hepatocellular carcinoma (HCC) and healthy control individuals, with notable upregulation in the HCC cohort (Fig. 1B) [7]. These findings support the potential of the signature genes identified in the biopsied tissues to serve as viable biomarkers for clinical applications. Furthermore, genes such as SPP1, which are intrinsically implicated in diseased tissues, have significant potential as critical markers for diagnosis. However, their applicability as biomarkers for non-invasive assessments may present limitations. Consequently, entities unearthed from multi-omics analysis of patient-derived tissue samples should undergo rigorous subsequent validation phases for their efficacious deployment as clinical biomarkers.
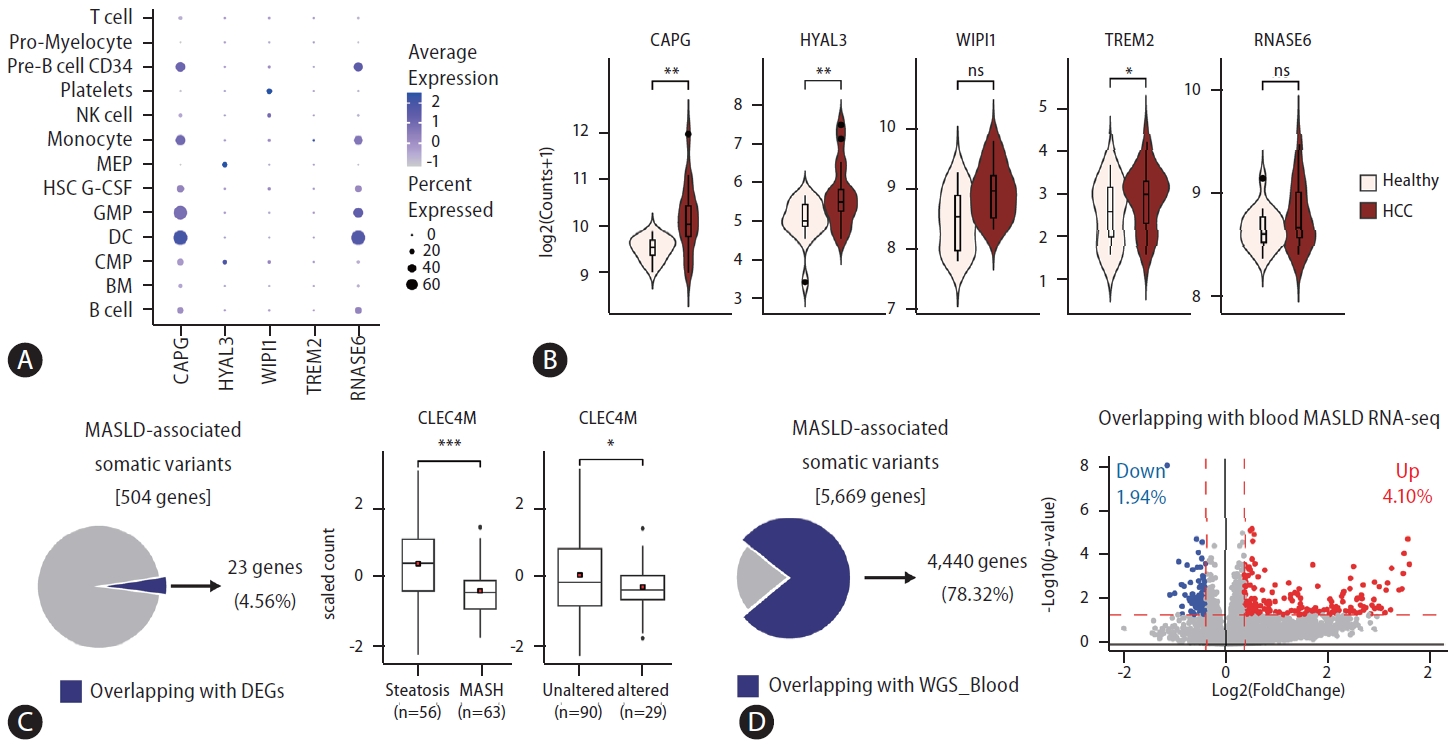
Applications of the signature gene set discovered in tissues of patients with metabolic-associated steatohepatitis liver disease (MASLD) for developing non-invasive diagnostic markers. (A) Investigation of the expression levels of signature genes in sub-cell populations from peripheral blood mononuclear cells in patients with MASLD through scRNA-seq analysis. Dot plot describing the expression levels of features in each cell type. (B) The expression levels of signature genes in liquid biopsy samples from both healthy individuals and patients with hepatocellular carcinoma (HCC). (C) Identification of genes with variations and changes in expression in MASLD progression. Box plot showing the expression level of CLEC4M grouped into pathology (middle) and altered status (right). (D) Identification of genes with detected variations in both patient tissues and blood samples. Scatter plot showing the differentially expressed genes between steatosis and metabolic dysfunction-associated steatohepatitis (MASH). Blue dots denote MASH-downregulated genes, and red dots denote MASH-upregulated genes. The red dashed line represents the cutoff value (horizontal: P-value*lt;0.05, vertical: |foldchange|>1.3). Pro-Myelocyte, progenitor myelocyte; Precursor B cell; NK, natural killer; MEP, megakaryocyte–erythroid progenitor cell; HSC, hematopoietic stem cell; GMP, granulocyte monocyte progenitors; DC, dendritic cells; CMP, common myeloid progenitors; BM, bone marrow. P-values are estimated using the Student’s t-test, P-value: *<0.05, **<0.01, ***<0.001.
Exploring MASLD-associated features, encompassing genomic variations, differentially methylated regions (DMRs), and differentially expressed genes (DEGs), from comprehensive omics datasets in previous studies requires thorough investigation. In a previous study, genomic variations, specifically somatic variations, were investigated, and approximately 5% of these variations (23/504 genes) were examined for changes in gene expression during disease progression (Fig. 1C). Among them, the gene expression levels of C-type lectin domain family 4 member M (CLEC4M), a gene involved in the immune response in the liver, substantially decreased in metabolic dysfunction-associated steatohepatitis compared to those in steatosis. Additionally, the decrease in gene expression was more pronounced in the altered group than in the unaltered group. Therefore, understanding the specific mechanisms and pathways of CLEC4M in MASLD is crucial for gaining insights into its potential as a therapeutic target or biomarker.
Building on this, we explored potential biomarkers by identifying genomic variations that were concurrently present in both the tissues and blood samples of patients with MASLD. Analysis of the WGS data obtained from tissue samples of patients revealed variants in approximately 5,700 genes, with 78% of these genes also present in the WGS data obtained from blood samples (Fig. 1D). Among these, 6% exhibited expression changes associated with MASLD progression [8]. To establish a direct link between these variations and changes in gene expression, further detailed investigations, such as eQTL analyses, are necessary. Nonetheless, the significance of our previous study lies in providing a consolidated dataset from a singular cohort, which offers a unique resource with substantial value for further research.
In conclusion, advancements in sequencing technologies have revealed an extensive repertoire of genetic and epigenetic determinants associated with MASLD. Nonetheless, rigorous validation of the pathogenic roles of these entities is imperative to identify key elements that influence disease etiology and therapeutic intervention and to ensure continuous commitment to advancing research and improving patient outcomes. Selecting signature gene sets with potential as biomarkers from a vast pool of MASLD-associated features, as achieved in previous research, conserves time required for experimental validation while maximizing the understanding of pathogenic ef fec ts during MASLD progression.
Notes
Authors’ contribution
S.J. and S.Y. contributed to the WGS, total RNA-seq, and scRNA-seq analyses; J.H.P., Y-S.L., and K.H.Y. conceived and designed the study; S.J., S.Y., and K.H.Y. drafted the manuscript; all authors have read and approved the manuscript.
Conflicts of Interest
The authors have no conflicts to disclose.
Acknowledgements
This research was supported by the Collaborative Genome Program for Fostering New Post-Genome Industry under the National Research Foundation (NRF) and funded by the Ministry of Science and ICT (MIST) (NRF-2017M3C9A6044519 to K.H.Y., NRF-2017M3C9A6044517 to Y-S.L., and 2022M3 A9B6017654 to K.H.Y. and J.H.P.).
Abbreviations
MASLD
metabolic-associated steatohepatitis liver disease
DC
dendritic cells
GMP
granulocyte monocyte progenitors
HCC
hepatocellular carcinoma
MASH
metabolic dysfunction-associated steatohepatitis
DMRs
differentially methylated regions
DEGs
differentially expressed genes