Liver sinusoidal endothelial cell: An important yet often overlooked player in the liver fibrosis
Article information

Abstract
Liver sinusoidal endothelial cells (LSECs) are liver-specific endothelial cells with the highest permeability than other mammalian endothelial cells, characterized by the presence of fenestrae on their surface, the absence of diaphragms and the lack of basement membrane. Located at the interface between blood and other liver cell types, LSECs mediate the exchange of substances between the blood and the Disse space, playing a crucial role in maintaining substance circulation and homeostasis of multicellular communication. As the initial responders to chronic liver injury, the abnormal LSEC activation not only changes their own physicochemical properties but also interrupts their communication with hepatic stellate cells and hepatocytes, which collectively aggravates the process of liver fibrosis. In this review, we have comprehensively updated the various pathways by which LSECs were involved in the initiation and aggravation of liver fibrosis, including but not limited to cellular phenotypic change, the induction of capillarization, decreased permeability and regulation of intercellular communications. Additionally, the intervention effects and latest regulatory mechanisms of anti-fibrotic drugs involved in each aspect have been summarized and discussed systematically. As we studied deeper into unraveling the intricate role of LSECs in the pathophysiology of liver fibrosis, we unveil a promising horizon that pave the way for enhanced patient outcomes.
INTRODUCTION
Liver fibrosis commonly occurs as a pathological reparative response to chronic liver diseases, manifesting as the excessive sedimentation or abnormal distribution of extracellular matrix (ECM) components, including collagen and glycoprotein in the space of Disse [1]. The majority of patients with liver fibrosis in the early stage have no obvious weaknesses or fatigue and don’t usually present with symptoms; however, as the disease progresses, symptoms such as pain, anorexia, sensitivity to fried foods and abdominal discomfort may gradually emerge and worsen to cirrhosis or hepatobiliary cancer [1], posing a significant threat to the global liver health. The main causes and underlying conditions associated with liver fibrosis include but are not limited to chronic hepatitis, long-term alcohol abuse, liver-overloaded lipids, accumulated bile acids, chemicals, metabolic disorders and genetic factors. As hepatic parenchymal cells of the liver, hepatocytes play a crucial role in the early stages of liver fibrosis by responding to liver damage, resulting in alterations in gene expression, immune responses, and hepatocyte apoptosis, making hepatocytes one of the focal cell types in the pathogenesis of liver fibrosis. The damaged hepatocytes caused by oxidative stress and endoplasmic reticulum (ER) stress secrete cytokines such as tumor necrosis factor-alpha (TNF-α), which recruit other immune cells and induce inflammatory responses [2]. The cumulative impacts of hepatocyte injury and regeneration, along with inflammation and the release of profibrotic factors triggered by immune cells, generate a fibrogenic milieu that additionally drives the activation of quiescent hepatic stellate cells (HSCs) into myofibroblast-like HSCs, commonly referred to as activated HSCs [3]. Activated HSCs subsequently secrete ECM components that contribute to the development of liver fibrosis and remodeling of the liver morphology, which involves the gradual replacement of the normal liver parenchyma by a fibrous scaffold composed of collagen fibers and other ECM proteins, ultimately leading to the compromised organ function, making activated HSCs another focal cell type responsible for the pathogenesis of liver fibrosis [4].
The overlooked impact of liver sinusoidal endothelial cells (LSECs) on liver fibrosis
Although the function of hepatocytes and HSCs in Disse space during the development of the liver fibrosis has been extensively explored [5], the advancements in therapeutic outcomes of liver fibrosis is unsatisfactory, which is possibly attributed to the restricted comprehension of LSECs. What makes LSECs potentially exert pivotal influence on the progression of liver fibrosis? Initially, the alterations in LSEC phenotypes and functions could actively participate in the development of liver fibrosis. Under physiological homeostasis, LSECs, as the predominant nonparenchymal cell type in the liver, not only establish a protective barrier in the hepatic sinusoid but also possess important physiological functions, such as material filtration, endocytosis, antigen presentation and leukocyte recruitment [6], making LSECs extensively engaged in maintaining the microenvironmental equilibrium. Throughout the progression of liver fibrosis, the function of LSECs subsequently affects and presents material communication barriers and reduces permeability, thereby influencing the filtering and clearing function of the liver [7]. The fenestrae of LSECs refer to the micropores or windows located on their surface, which are essential for facilitating the exchange of substances between the blood and Disse space [8]. Specifically, there is a reduction in the quantity and size of LSEC fenestrations; under certain profibrotic conditions, they may even vanish. Then, the reduction or loss of LSEC fenestrae depresses the efficiency of blood filtration in the hepatic sinusoids, suggesting that microorganisms, cell fragments and other harmful substances that resulted in liver injury and liver fibrosis in the blood may not be adequately eliminated, thus increasing the risk of liver damage ultimately [6]. Moreover, LSECs also participate in antigen presentation through fenestrae, thereby regulating the immune response. Novel clinical research from Ishikawa et al. [9] reported the expression level of the scavenger receptor FcγRIIb in LSECs contributed to the clearance of small immune complexes in hepatic sinusoids during liver fibrosis of nonalcoholic steatohepatitis (NASH). Besides, once defenestration of LSECs occurs, the immune regulatory function of LSECs is impaired, which further increase the susceptibility to immune-related liver diseases [10]. Simultaneously, there is an enhancement in intercellular connections and the formation of the basement membrane, collectively leading to a process termed sinusoidal capillarization. Then, the capillarized LSECs lose their characteristically high scavenging capabilities that safeguard efficient vasculature, acquiring vasoconstriction, pro-inflammatory, and prothrombotic functions
The ignored communication among LSECs and other hepatic cells in liver fibrosis
Given the significant functional role of LSECs in both physiological and pathological conditions, apart from the intrinsic changes of LSECs, more attention have been put on another crucial aspect. The ability of LSECs to mediate intercellular communication and molecular transfer significantly contributes to modulating disease progression by transmitting signals to hepatocytes or HSCs. In particular, hepatocytes play a critical role in the progression of liver fibrosis owing to their self-repair capacity. However, the significance of LSECs in this reparative mechanism is considerable and remains inadequately explored. LSECs have been reported to maintain intercellular communication and regulate signal transduction, thereby ensuring the coordination and effectiveness of hepatocytes in the repair process and governing liver fibrosis and regeneration. Mechanistically, the regulation of LSECs for hepatocytes are accomplished through multiple ways such as maintaining hemodynamic stability, stimulation of vascular proliferation, regulating immune response and preserving intercellular communication [11]. Specifically, LSECs not only uphold microenvironmental equilibrium, such as hemodynamic stability in the liver, to diminish inflammation and foster a more conducive milieu for hepatocellular self-repair, but also secrete growth factors and cytokines, such as hepatocyte growth factor (HGF) that could promote hepatocyte proliferation and repair to alleviate liver fibrosis. Also, the decline in LSEC fenestrae would affect the exchange of nutrients, hormones and other important substances between the blood and hepatocytes, resulting in metabolic disorders and impaired cell functions.
In addition to hepatocytes, as we previously mentioned, LSECs can also affect HSCs or modulate the molecular exchange with HSCs, collectively controlling the progression of liver fibrosis. Under normal physiological circumstances in the liver, differentiated LSECs maintains the quiescent state of HSCs, whereas persistent HSC activation contributes to the progression of liver fibrosis. Nonetheless, during liver injury, differentiated LSECs could inhibit HSC activation [12], promote their transformation into fibroblasts and could not effectively control the collagen deposition of HSCs either through indirect mechanisms such as paracrine secretion and mechanical signaling, or by direct contact with HSCs, which led to the overactivation of HSCs and ameliorated the progression of liver fibrosis [13]. Certain gaseous signaling molecules, such as nitrogen monoxide (NO) that contributes to uphold the vasodilation of hepatic sinusoids and reduce vascular tension, can be released by LSECs to balance the activation of HSCs in the Disse space [14]. Interestingly, fibronectin produced by LSECs can influence HSC phenotypes, thereby promoting their activation, which can further produce more ECM component to destroy the functional LSEC phenotypes in turn [15]. Hence, in contrast to the prevalent yet superficial hepatocytes and HSCs, with the permeability and ability to mediate substance exchange, LSECs actively participates in the cellular activities of multiple cell types during the development of liver fibrosis.
THE AIM OF THIS STUDY AND INVOLVED METHODS
Recently, a booming number of researches have focused on the function of LSECs in liver fibrosis. This comprehensive review not only aims to summarize the unique role and specific mechanisms of LSECs but also sheds light on the communication between LSECs and other cells in the development of liver fibrosis. Furthermore, various newly discovered natural compounds or molecules that exhibit potential in targeting LSECs are summarized and discussed, thereby emphasizing the promising applications of LSEC-targeted therapeutic drugs in the treatment of liver fibrosis. Here, we performed a literature review using the search terms “liver sinusoidal endothelial cell” and “liver fibrosis” in the PubMed database, limiting the search to articles published within the past decade with an impact factor equal to or greater than 3, by which we have collected 95 representative studies.
THE LSEC DEDIFFERENTIATION PROCESS IN LIVER FIBROSIS
LSEC fenestrae changes in the liver fibrosis
During LSEC dedifferentiation, LSEC fenestrae gradually decrease or vanish, resulting in reduced cell permeability and the formation of a continuous basement membrane between adjacent cells, ultimately resembling the configuration of continuous capillaries. Therefore, the loss of LSEC fenestrae constitutes the principal event defining LSEC capillarization [6]. It is reported that a natural compound, curcumol could alleviate the defenestration and downregulate the expression of basement membrane proteins including laminin and collagen type IV in leptin-induced LSECs, thereby attenuating the process of liver fibrosis. It was considered to be accomplished through the downregulation of urokinase plasminogen activator (uPA)/uPA receptor (uPAR) as well as the upregulation of matrix metallopeptidase 13 (MMP13) that responded for the treatment of curcumol [16]. Additionally, riociguat, a soluble guanylate cyclase stimulator, sustained the differentiation of LSEC and reinstated fenestration in liver fibrosis tissues, elevating the liver sinusoid permeability [17]. Furthermore, trimethylamine-N-oxide as gut microbial metabolite was found to increase LSEC fenestrations and reduced the basement membrane, thereby collectively preserving murine vascular function and mitigating the progression of liver fibrosis [18].
The cellular cytoskeleton includes microfilaments, microtubules, and intermediate filaments. Among them, the microfilament network formed by fibrous actin and microfilament-associated proteins on the cell membrane surface make the cell membrane strong and tough. Additionally, the microfilament network is highly involved in membrane deformation and maintaining cell shape, which are essential in the development of liver fibrosis [19,20]. It is reported that dynamic remodeling of the cytoskeleton and redox homeostasis are highly involved in the formation of fenestrations in LSECs. According to the reports, it has been demonstrated that bone morphogenetic protein 9 (BMP9) regulated LSEC fenestration, making it in a crucial paracrine regulator for maintaining liver homeostasisand hindering liver fibrosis [21,22]. In cell culture, it has been observed that LSECs underwent dedifferentiation and exhibited fenestrae loss, simulating a process reminiscent of capillarization. Notably, the dynamic cytoskeleton microfilament network was reported to be highly participated in the process. LSEC fenestrae were restructured by administering cytochalasin D (Cyto D), an F-actin-depolymerizing agent, which predominantly dispelled the formation of large stress fibers in LSEC dedifferentiation , leading to fenestrae reformation and consequently mitigating liver fibrosis [23]. Besides, the dynamic remodeling of the cellular cytoskeleton is intricately linked to cellular destiny, encompassing apoaptosis, aging and oxidative states [24]. Oxidative stress-induced injury mediated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 (NOX2) is also documented to exacerbate LSEC defenestration through premature senescence related to progestin. Sirtuin 1 (SIRT1), an essential protector resisting oxidative stress and senescence, mediated deacetylation of p53, which preserved LSEC defenestration and ameliorated liver fibrosis by suppressing premature senescence induced via oxidative stress and maintaining cytoskeleton [25]. In summary, we believe that targeting the dynamic remodeling process of the cytoskeleton might be a extremely promising research direction. Developing inhibitors that target the cytoskeletal microfilament network could potentially disrupt the mechanical LSEC integrity, thereby inhibiting the formation of fenestrations.
As liver fibrosis progresses, the porosity of LSECs gradually decreased, and their membrane permeability decreased. Vascular endothelial growth factor (VEGF) not only enhanced fibrogenesis but also facilitated liver tissue repair and fibrosis resolution by mediating sinusoidal permeability [26]. As reported, VEGF was reported to facilitate the engraftment of bone marrow mesenchymal stem cells (BMSCs), increase hepatocellular regeneration and ameliorate hepatic function [27], which mechanismically attributed to the upregulation of VCAM1 and increased LSEC permeability.
LSECs mediate liver fibrosis via controlling capillarization or angiogenesis
LSECs exhibited specific characteristics, including the absence of basement membrane proteins and incomplete tight junctions. In the initial phase of liver fibrosis, LSEC capillarized and dedifferentiated to lose their protective properties, showing as losing fenestrae, the unique ultra-microstructure of LSECs, forming organized basement membrane and enhanced tight junctions [28]. Meanwhile, LSECs can also stimulate neovascularization by secreting angiogenic factors, thereby fostering the fibrotic process. It is worth noting that LSEC capillarization precedes the activation of macrophages and HSCs in the process of liver fibrosis, making it a crutial process in the development of liver fibrosis. A previous clinical study conducted on patients with chronic hepatitis C confirmed that LSECs underwent only morphological alterations following hepatitis C virus infection, but in the initial stage of fibrosis, LSEC capillarization was observed, suggesting that LSEC vascularization may contribute to the development of liver fibrosis [29]. Specifically, Maretti-Mira et al. [30] suggested that the formation of capillarization is not attributed to the dedifferentiation of pre-existing LSECs in the liver, but rather to the incomplete LSEC differentiation derived from the bone marrow. Throughout the liver fibrosis process, these bone marrow-derived endothelial cells are recruited to the liver to repair damaged LSECs. However, in this process, they fail to completely differentiate into LSECs with typical fenestrations, leading to capillarization, which contradicts the previous theory that capillarization occurs in LSECs through the process of dedifferentiation, resulting in the loss of fenestrations. A recent study revealed that aging triggered LSEC capillarization, and the deficiency of endothelial C-kit could further disturb dynamic equilibrium in LSECs, along with enhancement of fibrosis and inflammation related factors expression including tissue inhibitor of metalloproteinases 1 (TIMP-1), collagen1, platelet-derived growth factor (PDGF)β, IL-6 and IL-1β, thus exacerbating inflammation and liver fibrosis in NASH [31].
Furthermore, in addition to LSEC capillarization, hepatic angiogenesis that defined as the formation of new blood vessels from pre-existing ones was also highly related with LSECs and contributed to the development of liver fibrosis [8]. CD31, VEGF, CD34 and tyrosine kinase with immunoglobulin like and EGF like domains 1 (Tie1) are considered as the detectable markers of angiogenesis. It is reported that heparinoid compound sulodexide downregulated the expression of CD31, CD34 and laminin [32] that have been proved to mediate LSEC capillarization, then further reversed the fibrotic progression of liver. Furthermore, CU06-1004, an endothelial dysfunction blocker, exerted the function of anti-fibrosis partially attributed to subduing hepatic sinusoidal capillarization through the downregulation of CD31 expression [33]. VEGF is a publicly known factor that highly associated with the LSEC capillarization and particularly, hepatic angiogenesis. Levels of VEGF and angiopoietin-2 in the serum of mice with liver fibrosis showed notable elevation [34]. Wang et al. [35] supported that the regulation of LSEC angiogenesis via VEGF played an important role in the process of liver fibrosis. Neuropilin-1 (NRP-1) is a complex transmembrane receptor that binds to both VEGF and vascular endothelial growth factor receptor 2 (VEGFR2) and is abundantly expressed in LSECs. Typically, when NRP-1 is overexpressed in LSECs, VEGFR2 is upregulated through the regulation of focal adhesion kinase and its corresponding kinase activity [35]. In addition, NRP1 potentiated VEGFR2-related angiogenesis via the phosphatidylinositol 3’-kinase (PI3K)/AKT pathway. Additionally, several agents have been documented to influence LSEC angiogenesis via modulating the protein function of VEGF. Leptin-stimulated LSECs exhibited the features of capillarization, which resulted from the increased expression of endothelin-1, VEGF, laminin and type IV collagen [36]. Otherwise, plumbagin was found to reverse the capillarization of hepatic sinusoids by downregulating the expression of these mRNA. Several studied have proved that tetramethylpyrazine (TMP), a natural anti-angiogenic ingredient originally isolated from Ligusticum wallichii, could ameliorate liver fibrosis both in vitro and in vivo. Zhao et al. [37] reported that the anti-fibrotic effects of TMP were achieved by mitigating LSEC capillarization via downregulating pro-angiogenic growth factors and their receptors, including VEGF-A, VEGFR2, PDGFβ and platelet-derived growth factor-receptor β (PDGF-Rβ) along with the angiogenic pathways that responded to these factors [37]. Additionally, the in vivo administration of levistilide A exhibited remarkable anti-fibrotic effects, which is evidenced by a reduction in collagen deposition and neovascularization [38]. Notably, this study attributed this phenomenon of anti-angiogenesis to the effects of levistilide A on preventing sinusoid capillarization through the downregulation of genes involved in the VEGF signaling pathway, including CD31, VEGF, and VEGFR2. Furthermore, aside from small molecular agents, biomacromolecules could also regulate VEGF signaling. Human recombinant endostatin, endostar, has been demonstrated to decrease the expression of VEGFR1 and VEGFR2, resulting in reduced LSEC capillarization [39]. In addition, the anti-angiogenic effects of clinical drugs that regulated VEGF-related signaling have been widely researched, demonstrating remarkable efficacy in anti-fibrosis. Vatalanib, a clinical VEGFR inhibitor, was identified to inhibit the mRNA expression of transforming growth factor (TGF-β), VEGFR1 and VEGFR2 and then altering the LSEC phenotypes, which decreased sinusoidal capillarization and ameliorated liver fibrosis [40]. Another clinical drug, lenvatinib could inhibit hepatic neovascularization and the expression of proangiogenic factors, including VEGF1, VEGF2, and VEGF-A, which collectively suppressed angiogenesis and ameliorated liver fibrosis [41]. Besides, administration of carvedilol hindered the development of liver fibrosis by depressing the expression of angiogenic factors such as VEGF and angiopoietin-2, leading to the suppression of sinusoidal capillarization and the changes in LSEC phenotypes [34]. Moreover, olmesartan treatment not only lessened the levels of VEGF, angiotensin II and PDGF, but also decreased the expression of their receptors such as VEGFR1, VEGFR2, and angiotensin II type 1 receptor (AT1R) [42] in liver fibrotic mice, thus blocking the angiotensin II-AT1R-VEGF axis, which resulted in anti-angiogenesis effects and improvement of hepatic sinusoidal remodeling.
In addition to VEGF, hypoxia inducible factor-1α (HIF-1α) is another common transcription factor that mediates the LSEC capillarization and angiogenesis, thus participating in the regulation of liver fibrosis. It was reported that miR-322/424 upregulated the expression of HIF-1α protein in LSECs by binding to the mRNA of Cullin 2, leading to its degradation and consequently exacerbating pathological angiogenesis [43]. In addition, numerous investigations have demonstrated that traditional Chinese medicine and natural compounds regulated LSEC capillarization and angiogenesis to ameliorate liver fibrosis by regulating HIF-1α and its associated pathways. Chinese medicinal formulation Fuzhenghuayu ameliorated LSEC capillarization, hepatic angiogenesis and angiogenesis-associated gens, such as CD31, VEGF, VEGFR2, p-ERK and HIF1α, and ultimately inhibiting hepatic fibrosis [44]. Meanwhile, Xuefuzhuyu decoction was also reported to attenuate liver fibrosis via anti-angiogenesis [45], which might be achieved by the lower VEGF level in LSECs induced by the suppressed HIF-1α expression. In addition, carthami flos extract (CFE), originating from edible herb carthami flos that was traditionally functioned in ameliorating blood circulation and modulating angiogenesis, was shown to not only surpress the platelet-derived growth factor receptor beta (PDGFRβ)/ERK/HIF-1α and VEGFR2/AKT/eNOS pathway, but also alleviate LSEC capillarization by modulating the expression of CD31, CD34 and vWF in liver fibrosis [46]. Notably, Zhang et al. [47] reported that hypoxia elevated the expression of VEGF-A and angiopoietin 2 (Ang2) in LSECs, which was reversed by oroxylin A, a natural active ingredient derived from Scutellariae radix. Mechanistically, oroxylin A downregulated the nuclear translocation of yes-associated protein (YAP), and subsequently suppressed the expression of target genes such as HIF-1α, VEGF-A and Ang2, ultimately leading to antiangiogenic effects [47]. The hedgehog (Hh) pathway was the upstream signaling of HIF-1α and regarded as a conserved morphogenic signaling, which could regulate LSEC capillarization and angiogenesis in a HIF-1α dependent or independent way. Hh signaling not only directly activated the transcription of HIF-1α, but also mediately influenced HIF-1α by activating PROX1 transcription, which subsequently control the protein stabilization of HIF-1α [48], thus accelerating LSEC angiogenesis. Notably, a natural compound, curcumol inhibited LSEC angiogenesis by inhibiting Hh signaling and the downstream HIF-1α expression. Subsequent research revealed that the increase in the expression of Hh signaling markers, including sonic hedgehog, patched-1, smoothened (SMO) and glioblastoma 1 (GLI1), which are stimulated by VEGF-A, was attenuated at both the mRNA and protein levels following the in vitro administration of TMP [49], indicating that disruption of Hh signaling plays a pivotal role in improving sinusoidal angiogenesis and inhibit LSEC capillarization via TMP. Another study reported that liver X receptors (LXRs), a subset of the nuclear receptors, governed classical Hh signaling and maintained LSEC differentiated phenotypes, resulting in the suppression of sinusoidal capillarization and angiogenesis [50].
Overactivation of Notch signaling promotes HSC activation, expedites collagen synthesis and cell proliferation, thereby accelerating the advancement of liver fibrosis. Recent studies have reported that proteins associated with the Notch signaling are highly involved in LSEC angiogenesis. In addition, a-kinase anchoring protein 12 (AKAP12), a scaffold protein, could inhibit LSEC angiogenesis, as interrupting the formation of basement membrane and suppressing endothelin-1 expression, leading to the fibrosis resolution [51]. Interestingly, the latest research findings indicate that delta-like ligand 4 (DLL4), a ligand related to the Notch pathway, plays a crucial role in preserving hepatic sinusoid homeostasis and is principally expressed in LSECs. Overexpression of DLL4 in LSECs has been shown to enhance hepatic sinusoidal capillarization and sinusoidal tension, thereby exacerbating liver fibrogenesis, which might be regulated by activating Notch signaling [52]. Gu et al. [53] discovered that miR-30c directly targeted DLL4 and subdued the downstream Notch pathway, leading to the suppression of proliferation, migration and angiogenesis ability of LSECs, which further regulated pathological angiogenesis in vivo and improved liver fibrogenesis. Therefore, it is suggested that DLL4 and other proteins related to Notch pathway may serve as a pivotal factor in angiogenesis and represent potential targets for therapeutic intervention in this process. Another research reported that leukocyte cell-derived chemotaxin 2 (LECT2), a functional ligand for Tie1 (a specific orphan receptor), exhibited anti-angiogenic properties both in vitro and in vivo by inducing Tie1 dephosphorylation, suppressing endothelial cell migration and neovascularization [54]. In addition, LECT2 exhibited multiple effects on hepatic vascular remodeling, inhibiting portal angiogenesis and promoting sinusoidal capillarization, consequently intensifying liver fibrogenesis.
Several studies have reported that when blood flows, shear stress is generated, which can be sensed and responded to by molecules on the cell surface and initial the signal transduction, resulting in the changes of behavior and function of LSECs, such as the control of nucleocytoplasmic translocation of transcription factors. Fluid shear stress can activate the expression of krüppel-like factors (KLFs), and facilitate their nuclear translocation, which could mediate LSEC angiogenesis, subsequently impacting the process of hepatic fibrosis [55]. A large number of articles have reported that the natural compound curcumol has demonstrated significant therapeutic effects on fibrosis through mediating the KLFs-related signaling. Gao et al. [56] demonstrated that, Krüppel-like factor 5 (KLF5) activation led to the upregulation of angiogenesis markers including CD31 and CD34, which was notably reversed by treatment with curcumol, further decreasing the production of mitochondrial reactive oxygen species (ROS), increasing the antioxidative stress response, and selectively suppressing extracellular-signal-regulated kinase (ERK) phosphorylation in a liver fibrosis mouse model. Another study found that curcumol restrained KLF5 protein and mRNA expression by suppressing autophagy and inducing p62 accumulation in LSECs in vitro, which further inhibited pathological angiogenesis of LSECs during liver fibrosis progression [57]. According to a recent study, KLF5 could influence the angiogenic characteristics of LSECs by modulating the glycolytic process, and regulating the expression of lactate dehydrogenase-A (LDH-A) by transcriptionally combining with its promoter. Meanwhile, LDH-A was found to possess a non-enzymatic role within the nucleus, where it interacts with KLF5 to form a transcriptional complex, further potentiating KLF5 activity and resulting in a positive feedback loop [58]. Additionally, this KLF5/LDH-A feedback loop could be disrupted following the administration of curcumol that downregulated glycolytic enzymes induced by KLF5 overexpression in mRNA levels and decreased the intimate connection of KLF5 and LDH-A, thus collectively inhibiting the angiogenic properties of LSECs and attenuating liver fibrosis. During liver fibrosis, simvastatin promoted the upregulation of KLF2 and eNOS expression in LSECs [59]. Interestingly, in response to the abnormal fluid shear stress happened in liver fibrosis, KLFs were activated, exacerbating the dysfunction of LSECs, thereby representing potential targets for inhibiting the progression of liver fibrosis. The summarized pathways that we mentioned above were shown in Figure 1.
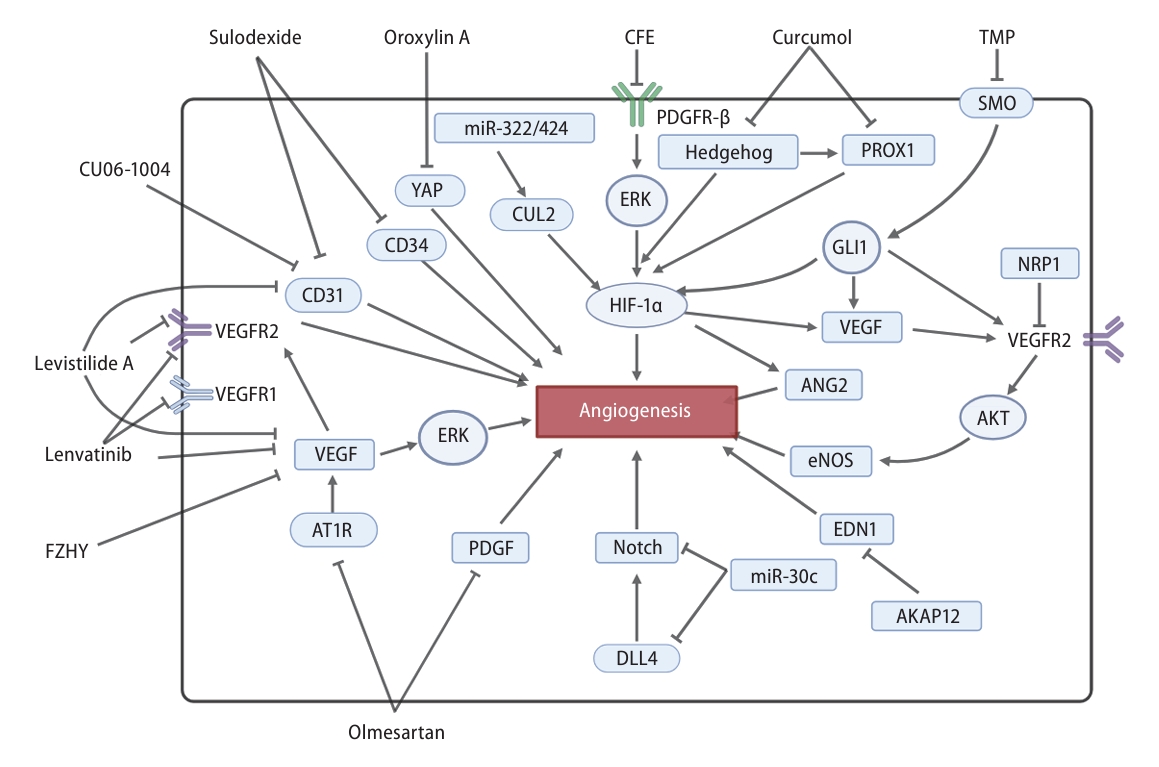
Mechanisms of LSEC angiogenesis and the related pharmacological intervention in the process of liver fibrosis. VEGF and HIF-1α were the two most crucial pathways in the process of LSEC angiogenesis. Treatments such as lenvatinib, levistilide A, olmesartan, TMP and FZHY could inhibit the VEGF pathway, exerting anti-angiogenic effects. Oroxylin A, CFE, curcumol and TMP could reverse the activation of angiogenesis caused by the HIF-1α pathway. Angiogenesis mediated by DLL4/Notch pathway was suppressed by miR-30c. AKAP12 inhibited EDN1-mediated angiogenesis. Olmesartan inhibited PDGF-mediated angiogenesis. Levistilide A, CU06-1004, and sulodexide could reduce LSEC angiogenesis by inhibiting the expression of CD31 or CD34. LSEC, liver sinusoidal endothelial cell; VEGF, vascular endothelial growth factor; HIF-1α, hypoxia inducible factor-1α; TMP, tetramethylpyrazine; DLL4, delta-like ligand 4; PDGF, platelet-derived growth factor.
Other phenotype changes or transition of LSECs in liver fibrosis
In addition to alterations in LSEC morphology, such as defenestration, LSECs, as one of the most vulnerable liver cell types after liver injury, not only exhibit multiple phenotypic changes, including characteristic changes or transitions to an inflammatory phenotype but also undergo a series of changes to acquire a mesenchymal phenotype, collectively resulting in loss of the ability to maintain liver homeostasis. Brougham-Cook et al. [60] conducted numerous in vitro experiments to explore alterations of LSEC phenotypes in diverse liver fibrosis microenvironments. They found that microenvironmental factors such as stiffness, ECM and soluble factors profoundly influenced the expression of LSEC phenotype markers, including Lyve-1, VE-cadherin, and CD31, which symbolized the changes of LSEC phenotypes. Following lanifibranor treatment, an agonist of pan-peroxisome proliferator-activated receptor (pan-PPAR), improvements in fenestration and alleviation of capillarization in LSECs were observed, along with a simultaneous decrease in the expression of VCAM1, intercellular adhesion molecule 1 (ICAM1), and E-selectin in LSECs, which indicated an improvement in LSEC phenotype and a reduction in pro-inflammatory phenotype [61]. In summary, lanifibranor reduced hepatic vascular resistance and improved microvascular functions and the LSEC phenotypes, thereby alleviating liver inflammation and leading to the regression of liver fibrosis.
However, as we have mentioned, the LSEC phenotype is frequently variable and can undergo dynamic transitions, including processes like endothelial-mesenchymal transition (EndMT). When LSECs undergo this process, they lose specific endothelial cell functions and phenotypes, while acquiring the characteristics of mesenchymal cells. Ruan et al. [62] discovered that during the process of liver fibrosis, capillarized LSECs underwent partial EndMT, resulting in the redundant production of ECM components such as collagen and other fibrosis-related proteins produced by LSECs, which further accumulated in the liver sinusoids, thus promoting liver fibrosis. In this process, LSECs experienced a continuous phenotypic alteration, transitioning from normal cell dedifferentiation to capillarization and subsequently to EndMT. Hammoutene et al. [63] reported that the impairment of autophagy in metabolic dysfunction-associated steatotic liver disease (MASLD) could lead to the promotion of EndMT in LSECs, as well as induce cellular inflammation and apoptosis, ultimately facilitating the formation of liver fibrosis. Notably, the underlying mechanisms of this process have gradually been revealed. Notably, the transcriptional regulator megakaryocytic leukemia 1 (MKL1) and signal transducer and activator of transcription 3 (STAT3) collaborate to stimulate the transcription of Twist1, thereby amplifying TGF-β induced EndMT in LSECs and worsening liver fibrosis [64]. It has been reported that brahma-related gene 1 (BRG1) could regulate the production of ROS and EndMT in endothelial cells both in vivo and in vitro, which depended on the production of NADPH oxidase 4 (NOX4), suggesting that targeting BRG1-NOX4 axis might inhibit the development of liver fibrosis via blocking EndMT process [65]. A recent study has demonstrated that elevated serum level of high-mobility group box 1 (HMGB1) could induce EndMT in LSECs. This EndMT process led to heightened ECM production, ultimately resulting in a diminished capacity of LSECs to inhibit HSC activation during liver fibrosis [66]. Moreover, the administration of the anti-fibrosis natural compound silymarin was reported to reduce serum HMGB1 levels and suppress EndMT via promoting the expression of Egr1 that could alleviate liver fibrosis by inhibiting HMGB1. Similarly, the natural polyphenolic compound chlorogenic acid (CGA) not only reduced the release of HMGB1 in LSECs but also decreased ECM deposition in the liver sinusoids and HMGB1-induced ECM production, thus alleviating liver fibrosis in NASH [67].
REMODELING OF INTRACELLULAR PATHWAYS FOR LSEC HOMEOSTASIS BALANCE
The vascular secretion signals of LSECs regulate liver fibrosis
Vascular secretory signals, which were predominantly generated by the majority of endothelial cells, play a crucial role in regulating vascular remodeling, inflammatory responses, and fibrotic processes. Dysregulation of these genes can exacerbate liver damage and lead to functional impairments, underscoring their significance in the progression of liver fibrosis [68]. Similar to endothelial cells elsewhere, LSECs also produce the different vascular secretion signals during acute and chronic liver injuries, giving rise to two completely different pathological processes: liver regeneration or fibrosis. With acute liver injury, LSECs upregulated the expression of CXCR7 and activated the transcription factor ID1, subsequently promoting liver regenerative vascular niche through the secretion of pro-regenerative vascular factors. However, with chronic liver injury, the activation of fibroblast growth factor receptor 1 (FGFR1) induced upregulation of CXCR4 expression in LSECs, which further suppressed the pro-regenerative CXCR7-ID1 pathway and accelerated liver fibrosis, indicating the dominance of the FGFR1-CXCR4 vascular secretion pathway [69,70]. Serving as essential ligands for chemotactic receptors, numerous chemokines participate in both vascular secretion and the liver fibrosis process of LSECs. During early CXCL1-mediated liver fibrosis, glycolysis enhanced the vascular secretion processes involving CXCL1 in LSECs, which was attributed to glycolysis promoting CXCL1 expression via the movement of NF-kB and its interaction with actin polymerization, thereby promoting the progression of liver fibrosis [71]. In the liver, GATA4 functioned as a developmentally-induced transcription factor intricately linked to liver development and regeneration. Depletion of GATA4 in liver triggered HSC activation and accelerated the process of liver fibrosis [72]. According to a recent study, GATA4 deficiency in LSECs could induce sinusoidal capillarization in liver fibrosis, leading to the re-expression of fibrogenic vascular secretory factors including PDGFβ, SPARCL1, ESM1, and IGFBP5. The endothelial transcriptional regulator MYC, which is mediated by GATA4, was activated and functioned as a downstream driver of liver fibrosis along with the vascular secretory growth factors PDGFβ and PDGFRβ. In summary, the GATA4/MYC/PDGFβ/PDGFRβ axis associated with vascular secretory signaling could regulate liver fibrosis, making it a potential therapeutic target [73]. In addition to transcriptional regulation, epigenetic modification was identified as another pathway through which these exogenous substances stimulate LSECs. As a transcriptional coactivator, P300 could modify histone proteins and promote gene activation, which was reported to accelerate the process of liver fibrosis. Besides, P300 could upregulate the expression of C-C motif chemokine ligand 2 (CCL2) via interacting with nuclear factor kappa B (NF-κB) and bromodomain containing 4 (BRD4) in LSECs after liver injury, thus stimulating LSECs to release the vascular secretion signals associated with liver diseases and exacerbating liver fibrosis [74].
A noteworthy star signaling: the NO pathway of LSECs regulates liver fibrosis
Publicly acknowledged, gas signaling molecules play important roles in various physiological and pathological processes, encompassing vascular regulation, cellular apoptosis, and inflammatory responses, among others. NO, as a second messenger molecule with high free radical activity, plays a crucial role in the maintenance of vascular tone and the regulation of blood pressure, which was produced by eNOS (endothelial nitric oxide synthase) in endothelial cells. Reportedly, activation of Notch in endothelial cells altered the vascular secretion function of LSECs and inhibited the eNOS-sGC pathway to downregulate Wnt2a and Wnt9b that are publicly known as hepatocyte mitogens to promote hepatocellular regeneration, subsequently accelerating LSEC dedifferentiation and exacerbating liver fibrosis [75]. Shao et al. [76] reported that BRG1 suppressed eNOS activity and reduced the bioavailability of NO in LSECs by regulating the vascular secretion signaling of LSECs, contributing to the worsening of liver fibrosis. Similarly, Jiang et al. [77] reported that the infection with schistosoma japonicum resulted in decreased differentiation and increased LSEC dedifferentiation, along with reduced NO secretion and enhanced TGF-β secretion. Meanwhile, several chemical and natural compounds have shown great anti-fibrosis effects via modulating the NO signaling. Human placental extract (HPE) has been reported to enhance the vitality of LSECs through increased eNOS expression, while also improving serum AST and ALT levels, indicating the amelioration of liver fibrosis in NASH [78]. After the administration of emricasan, the LSEC phenotypes changed, such as the restoration of endothelial fenestration and increased bioavailability of NO, which further led to the activation of eNOS and cGMP enhancement, ultimately alleviating liver fibrosis [79]. In addition, a natural polyphenolic flavonoid resveratrol possesses the potential to enhance hepatic vasorelaxation and endothelial dysfunction and increase NO bioavailability in LSECs, while decrease thromboxane A2 production, downregulate the expression of TGF-β and NF-κB and reduce desmin and a-SMA protein expression, ultimately attenuating liver fibrosis [80].
NO and ROS are balanced under physiological circumstances, a critical aspect for maintaining homeostasis in LSECs. ROS can scavenge NO by superoxide (O2-), resulting in the reduced NO bioavailability, thus promoting the development of liver fibrosis. After liver injury, there was a gradual transition from a pro-regenerative phenotype to a pro-fibrotic phenotype in LSECs, which influenced the process of liver fibrosis through the ERK1/2-AKT axis [81]. Mechanistically, ERK1/2 shifted the equilibrium between NO and ROS towards NO, thus maintaining LSEC homeostasis and promoting regeneration. On the other hand, AKT could tilt the balance towards ROS, resulting in LSEC dysfunction and promoting liver fibrosis. Zheng et al. [82] provided additional evidence that curcumol regulates hepatic angiogenesis by hindering the upstream signaling pathway of NO and subsequently modulating NO expression. They also found that curcumol could downregulate the VEGF/AKT/eNOS pathway, leading to the enhancing fenestration of sinusoidal endothelial cells and attenuating liver angiogenesis. Additionally, another mechanism by which NO modulates the ROS process during liver fibrosis may be involved in the induction of autophagy. Autophagy in endothelial cells contributed to preserving the dynamic equilibrium of LSECs by enhancing the bioavailability of NO and eliminating accumulated ROS, which maintained LSEC phenotype, reduced oxidative stress, and ultimately reversed liver fibrosis [83]. However, this phenomenon was predominantly observed during the initial phases of liver injury, but in late-stage chronic damage, autophagy was insufficient to reverse fibrosis. Previous studies have reported conflicting roles of autophagy in liver fibrosis [84]. The autophagic activity in LSECs also exhibits opposing effects on the progression of liver fibrosis. Autophagic degradation of Caveolin-1 (Cav-1) and remodeling of F-actin were found to promote the LSEC defenestration and the development of CCL4-induced liver fibrosis by suppressing the NO-dependent PI3K-Akt-mTOR pathway [85]. Crucially, whether promoting or inhibiting autophagy, both exert their effects on liver fibrosis by intervening the downstream NO signaling pathways. Similar with NO, carbon monoxide (CO) act as antagonists of endothelin-1 by mediating relaxation of sinusoidal vessels [86], but the function of CO in LSEC and the intercellular communication involving LSECs for liver fibrosis still not well-studied, which was worthy for further investigation.
INTERCELLULAR COMMUNICATION INVOLVING LSECS AND ITS IMPLICATIONS IN LIVER FIBROSIS
LSECs indirectly mediate liver fibrosis via hepatocytes
The reciprocal interaction between hepatocytes and LSECs is pivotal for maintaining normal liver physiological function and structure. LSECs, on one hand, safeguard hepatocytes by regulating the permeability of the sinusoidal capillaries, while on the other hand, they activated hepatocyte proliferation through inhibiting the expression of hepatocyte growth inhibitor TGF-β and promoting the secretion of growth factors. It was reported that growth factors, including but not limited to BMP2 and BMP6, were secreted by LSECs in a manner of paracrine to hepatocytes, regulating hepcidin-dependent iron homeostasis and fibrotic processes [87]. TAZ-knockout in LSECs could decrease NO production and interrupt the normal regeneration process after partial hepatectomy, which subsequently lead to aberrant transport of cellular signaling for hepatocytes, ultimately promoting the development of liver fibrosis [14]. Substance P, a neuropeptide expressed in numerous tissues, have been shown to ameliorate tumor necrosis factor (TNF-α) induced endothelial dysfunction, in numerous and promote LSECs to release HGF, which promoted hepatocellular regeneration in a paracrine manner [88].
It has been reported that during hepatocellular damage process, LSECs exhibited reduced scavenger function, a phenomenon induced by the inflammatory factors and other cytokines released by hepatocytes. Reportedly, CD147 as glycoprotein was found to ameliorate the progression of liver fibrosis through mutual communication between hepatocytes and LSECs. Mechanistically, the overexpression of CD147 promoted AKT phosphorylation and activated the PI3K/AKT pathway, which stimulated hepatocytes to produce VEGF-A that induced the expression of VEGFR-2 in LSECs, thus stimulating angiogenesis and exacerbating liver fibrosis [89]. In addition, in the context of liver injury, damaged hepatocytes secreted semaphorin 3E, which triggered the contraction of LSECs and orchestrates liver sinusoidal regeneration, thus facilitating wound healing [90]. Simultaneously, during the reconstruction of sinusoid structures, semaphorin 3E overexpression in denatured hepatocytes could activate HSCs in LSEC-mediated manner, resulting in the progression of liver fibrosis process. Given the critical function of HMGB1 in preserving LSEC phenotypes and its recognized capacity to influence cell fate, we also detected the effects of HMGB1 on ages LSECs and liver damage. Our study reported that oxidative hepatocytes highly expressed and released HMGB1 into the liver microenvironment, which were further taken up and involved in LSEC fenestration and vascularization, but luckily, was inhibited by acteoside, a small molecular from Rehmanniae Radix Praeparata [91]. Thus, it is worthwhile to explore whether and how the HMGB1 secreted by hepatocytes intervened in the process of liver fibrosis by modulating LSEC functions.
LSECs indirectly mediate liver fibrosis via HSCs
During liver injury, the capacity of LSECs to prevent HSC activation is compromised or diminished, attributable to the LSEC dedifferentiation and LSEC capillarization [13]. Multiple signaling pathways altered in LSECs could impact HSC activation, primarily through the paracrine mediation of secretory factors. Reportedly, LSECs-derived adipocyte fatty acid binding protein (A-FABP) exacerbated liver fibrosis in mice by potentiating LSEC capillarization and activating HSCs, which was mechanistically attributed to the stimulation of the Hh signaling and the compromised inhibitory effect of LSECs on HSC activation. A-FABP originating from LSECs also influenced HSCs in a paracrine manner, which enhanced the transactivation of TGF-β by regulating c-Jun N-terminal kinase (JNK)/c-Jun signaling in HSCs, subsequently aggravating liver fibrosis. Additionally, plasma kallikrein (PLK) within LSECs has been shown to promote the activation of TGF-β, leading to the activation of HSCs and promoting liver fibrosis [92]. According to a recent report, downregulation of HIF-1α or CXCR4 alleviated the LSEC dedifferentiation and suppressed HSC activation, which can partially be attributed to the reduced release of PDGF-BB when LSEC dedifferentiation was inhibited, leading to the downregulation of PDGFR-β in HSCs [93]. While elevated expression of CXCR4 resulted in increased PDGF-BB expression in LSECs. PDGF-BB bound to its receptor PDGFR-β on LSECs, inhibiting the expression of CXCR7 and promoting LSEC dedifferentiation, which subsequently leads to HSC activation. In general, the HIF-1α/CXCR4/PDGF-BB/CXCR7 axis could regulate the LSEC dedifferentiation, thereby mediating HSC activation and liver fibrosis. For transcriptional regulation, silencing a transcription factor associated with LSECs, Zeb2 suppressed the expression of secretory factors including Gdf15, Igf1, and Ltf, in LSECs, which functioned as ligands that could activated the target genes in HSCs, thus mitigating the progression of liver fibrosis [94]. Furthermore, a novel cellar communication mechanism between LSECs and HSCs was discovered, which was regulated by long non-coding RNA (lncRNA). Chen et al. [95] discovered that lncRNA Airn can sustain LSEC differentiation both in vitro and in vivo, subsequently suppressing HSC activation and stimulating hepatocyte proliferation by upregulating the expression of Wnt2a and HGF through the paracrine pathway of LSECs, thus improving liver fibrosis. The mechanism can be summarized as the interaction between Airn and EZH2, which maintained LSEC differentiation via the KLF2-eNOS-sGC pathway, thus maintaining HSC quiescence and boosting hepatocyte proliferation [95].
Besides, mechanical signal transduction and vesicle transport are also involved in the transport of substances between LSECs and HSCs. Liu et al. [96] established fibrotic microniches (FμNs) that consisted of two-dimensional cultured LSECs and HSCs covered with three-dimensional collagen hydrogel to explore the regulatory of LSECs in HSC activation. Mechanistically, in the initial stage of liver fibrosis, LSEC angiogenesis is mechanistically linked to the condensation of collagen fibrils. These compacted collagen fibers then act as mechanical transducers, transmitting signals to HSCs through the discoidin domain receptor 2 (DDR2)-JAK2/PI3K/AKT-myocardin pathway, ultimately promoting the development of liver fibrosis. Chen et al. [97] conducted research examining the influence of autophagy on LSECs during hyperaldosteronism. They found that aldosterone-induced autophagy promoted the degradation of multivesicular bodies in LSECs, resulting in a reduction in the quantity and quality of extracellular vesicles transferred from LSECs to HSCs, which were responsible for maintaining the quiescence of HSCs. Consequently, HSCs were activated, culminating in the progression of liver fibrosis in the context of hyperaldosteronism [97]. Liao et al. [98] reported that the overexpression of S1pr2 in LSECs regulated the phosphorylation of YAP, leading to the activation the expression of TGF-β and excessive production of excessive ECM, which ultimately enhancing HSC activation and exacerbating liver fibrosis. VCAM1 derived from LSECs also mediated the activation of HSCs and promoted the development of liver fibrosis by activating the YAP1 pathway in HSCs [99]. Although there is no report indicating that the SIPR2 could be packaged in exosomes and transported from LSECs to HSCs at present, it has been demonstrated that SPHK1, which shares a similar function with SIPR2 as regulating the generation of SIP, has been confirmed to be transported within exosomes [100]. Additionally, exosomal sphingosine kinase 1 (SphK1) derived from LSECs has been identified as crucial in facilitating HSC migration, activation, and AKT phosphorylation, which further sustained HSC activation. The in vitro experiments demonstrated that the phenolic compound, salidroside, could alleviate this pathological process by inhibiting AKT phosphorylation, thereby reducing liver fibrosis [100]. Several agents have been reported to exhibit antifibrotic effects on communication between LSECs and HSCs. A prior investigation demonstrated that Scutellariae radix downregulated the expression of monocyte chemotactic protein 1 (MCP-1) to inhibit lipopolysaccharide-induced activation of LSECs, thereby suppressing HSC migration and ultimately alleviating liver fibrosis [101].
On the other hand, LSECs possess the ability to activate HSCs through direct contact. Several studies have reported the involvement of junctional adhesion molecules (JAM) in the interactions between LSECs and HSCs in liver fibrosis. Brozat et al. [102] demonstrated that JAM-A played a key role in maintaining the structural integrity of LSECs and preventing capillarization, which might keep the quiescence of HSCs through LSECs-derived paracrine mechanism, thus attenuating hepatic fibrogenesis. In addition, the expression of classical JAM including JAM-B and JAM-C, elevated throughout the progression of liver fibrosis. After activation, HSCs produced JAM-C, which allows activated HSCs as pericytes to connect with JAM-B-expressing LSECs, facilitating the interaction between the two cell types [103]. Simultaneously, JAM-B and JAM-C exhibited heterophilic interactions, thereby modulating the interplay between LSECs and HSCs, ultimately controlling liver fibrosis. A recent study showed that IVA phospholipase A2 (IVA-PLA2), a key isozyme of PLA2s in mammals, expressed in LSECs may promote sinusoidal capillarization in the liver, further leading to HSC activation and ensuing collagen deposition, which ultimately accelerated the process of liver fibrosis [104].
LSECs interact with other cells in liver fibrosis
In the preceding section, we discussed the effect of LSECs on hepatocytes and HSCs (Fig. 2), then we will delve into how could other cell types mediate liver fibrosis by influencing LSECs (Fig. 3). T helper cells and Kupffer cells (KCs) play crucial roles in the initiation and aggravation of liver fibrosis via modulating immune responses, inflammatory reactions and the release of cytokines. Zhong et al. [105] reported auxiliary T helper (Th) 1 and Th2 cells interacted with LSECs in vivo though different adhesion molecule to exert contrary effects on the process of liver fibrosis. The interaction between Th1 cells and LSECs could potentially expedite the reduction of LSEC fenestrae via the Rho-ROCK-myosin pathway, promoted LSEC angiogenesis, and ultimately exacerbating liver fibrosis, while the interaction between Th2 cells and LSECs exhibited the opposite tendency. During the process of liver fibrosis, LSECs could function as antigen-presenting cells that could recruit KCs by controlling the secretion of different matrix metalloproteases (MMPs) and tissue inhibitors of MMPs (TIMPs), which have been summarized in the previous review [15]. Recent studies have delved deeper into unraveling the mechanisms underlying the communication between LSECs and KCs. It is well known that KCs are tissue-resident macrophages located in hepatic sinusoidal cavities, making them a representative cell type of the liver. Sakai et al. [106] found that the Notch ligand DLL4 in LSECs was highly expressed and connected with the corresponding receptors Notch1 or Notch2 in bone marrow (BM) progenitor cells to promote a transition of the gene expression profile toward that of KCs. Moreover, the ligands including the TGF-β family ligands, BMP2, DLL4 and endogenous LXR from LSECs were found to be highly expressed and connected with their corresponding receptors such as TGFβR, Notch1, Notch2, LXR in KCs, which was required for the maintenance of Kupffer cell identity [106]. Likewise, the levels of integrins such as α4β1, αLβ2, αMβ2, and αXβ2 in KCs were increased while the expression of integrin ligands including Icam-1 and Vcam-1 that have the binding properties with these integrins were also increased, indicating that the integrins and integrin ligands were possibly participant in the direct adhesion process between KCs and LSECs [107]. Furthermore, in severe liver injuries, it has been shown through various studies that LSECs release proinflammatory cytokines, including TNF-α, IL-6, IL-1, and CCL2, thereby activating KCs. In turn, when LSECs was cocultured with Kupffer cell, LSECs also exhibited pathological phenotype changes even in the suitable well-maintained elasticity of matrix [108], which indicated that activated KCs impair the normal LSEC functions. Additionally, another study has reached similar conclusions that LSEC dedifferentiation has been observed in an inflammatory microenvironment created by KCs, suggesting that the co-culture of LSECs and KCs is likely not conducive to maintenance of LSECs [109].
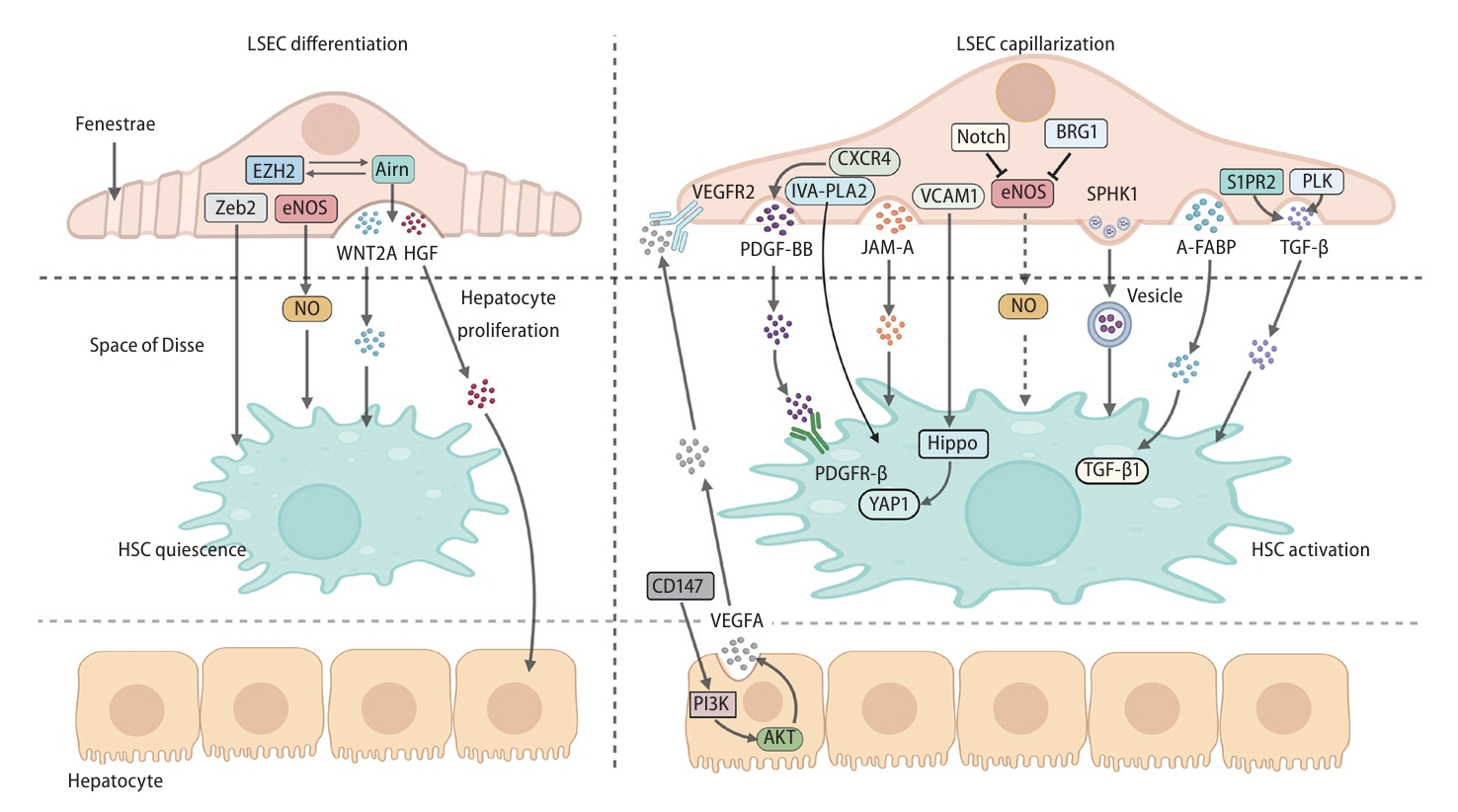
Communication among LSECs, HSCs and hepatocytes around the space of Disse during liver fibrosis. The expression of Zeb2 and eNOS in LSECs maintained HSC quiescence. Arin interacted with EZH2 to maintain HSC quiescence and hepatocyte proliferation through releasing WNT2A and HGF in paracrine manners. When stimulated by CD147, hepatocytes promoted LSEC capillarization through the PI3K/AKT/ VEGF-A/VEGFR2 axis. PDGF-BB stimulated by CXCR4, TGF-β stimulated by S1PR2 or PLK, JAM-A and A-FABP and were released from LSECs in paracrine manners and then activated HSCs. Exosomal SPHK1 from LSECs activated HSCs through vesicle transport. VCAM1 derived from LSECs stimulates HSC activation through Hippo and YAP1 pathways. Notch and BRG1 inhibit eNOS, reducing the bioavailability of NO and losing the ability to suppress HSCs. A-FABP, adipocyte fatty acid binding protein; AKT, AKT serine/threonine kinase; BRG1, brahma-related gene 1; CXCR4, C-X-C chemokine receptor 4; eNOS, endothelial nitric oxide synthase; EZH2, enhancer of zeste homolog 2; HGF, hepatocyte growth factor; HSC, hepatic stellate cell; IVA-PLA2, group IVA phospholipase A2; JAM-A, junctional adhesion molecule A; LSEC, liver sinoidal endothelial cell; NO, nitric oxide; PDGF-BB, platelet-derived growth factor-BB; PDGFR-β, platelet-derived growth factor receptor β; PI3K, phosphoinositide 3-kinases; PLK, polo-like kinase; S1PR2, sphingosine-1-phosphate receptor 2; SPHK1, sphingosine kinase 1; TGF-β, transforming growth factor beta; VCAM1, vascular cell adhesion molecule 1; VEGFA, vascular endothelial growth factor A; VEGFR2, vascular endothelial growth factor receptor 2; WNT2A, WNT family member 2a; YAP1, yes-associated protein 1.
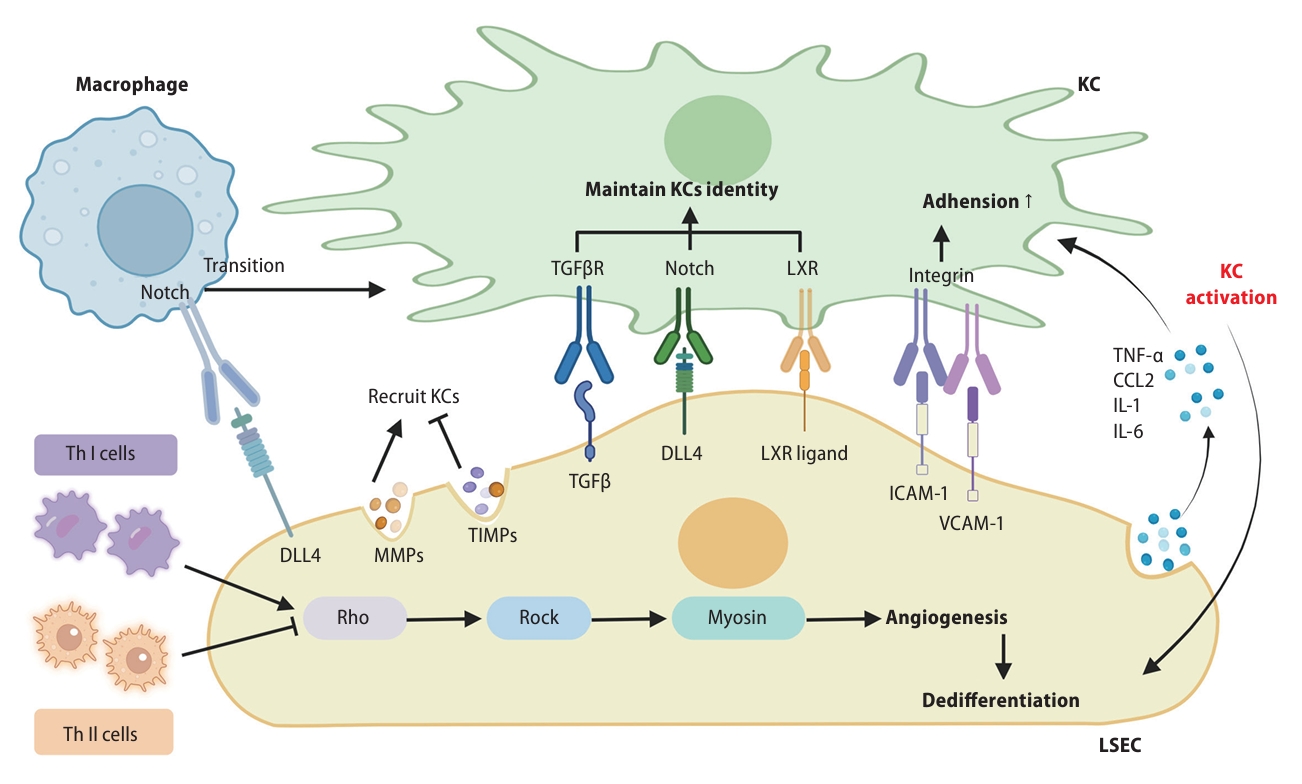
The interaction among LSECs with other cells. LSECs promote the transformation of macrophages into KCs through the binding of DLL4 to Notch receptor in macrophage. LSECs regulate the recruitment of KCs by secreting MMPs and TIMPs. The ligands such as ICAM-1 and VCAM-1 in KCs bind with the integrins on KCs and participate in the direct adhesion process between KCs and LSECs. TGF-β, DLL4, and LXR ligand in LSECs can interact with receptors such as TGFβR, Notch, and LXR in KCs, then maintaining the identity of KCs. Moreover, LSECs can secrete pro-inflammatory mediators to activate KCs. Th 1 and Th2 cells promote or inhibit angiogenesis in LSECs through the Rho-ROCK-myosin pathway. LSEC, liver sinusoidal endothelial cell; DLL4, delta-like ligand 4; VCAM1, vascular cell adhesion protein 1; TGF, transforming growth factor.
FUTURE EXPECTATION
This review provides a detailed description of the alterations that occurs in LSECs during the process of liver fibrosis and how these changes subsequently mediate liver fibrosis through distant pathways (Fig. 4). At the same time, an overview was provided on the therapeutic benefits of diverse clinical drug, small molecular agent, biomacromolecule, Chinese medicinal formulae and Chinese medicinal herb in addressing liver fibrosis by targeting LSECs (Table 1). Consequently, LSECs, which have received limited attention, play an important role in the pathogenesis of liver fibrosis, and may hold great promise as a prospective avenue for future development and therapeutic interventions in the treatment of liver fibrosis. It is publicly known that liver fibrosis is intricately linked to metabolic dysfunction-related fatty liver disease (MASLD) and viral hepatitis. In MASLD, with the increase of hepatic angiogenesis, pathological LSECs not only release inflammatory mediators and promote the recruitment of inflammatory cells, but also lose their ability to maintain HSC quiescence and release fibrogenic mediators, leading to the formation of liver inflammation and fibrosis collectively. Hence, enhancing and reinstating the functionality of LSECs may be an effective treatment strategy to prevent the progression of MASLD and ameliorate complications [110]. It has been reported that in the process of HCV infection, bone morphogenetic protein 4 (BMP4) secreted from LSECs could further promote HCV replication in hepatocytes and ameliorate the process of liver fibrosis [111], indicating that interrupting LSEC functions might potentially prevent the progression of HCV-induced hepatitis towards liver fibrosis. Besides, liver fibrosis is often accompanied by the process of liver regeneration, and both of these pathological processes are closely associated with LSEC capillarization or angiogenesis. During the initial induction phase of liver regeneration, the decrease in Ang2 expression resulted in reduced TGF-β production in LSECs, enabling hepatocyte proliferation by releasing an angiocrine signal. In the subsequent angiogenic phase of liver regeneration, the expression of Ang2 restored in LSECs, and the expression of VEGFR2 was further regulated to achieve regenerative angiogenesis. This research showed that LSECs functioned as a dynamic regulator of liver regeneration, potentially contributing to regeneration induced by fibrosis [112].
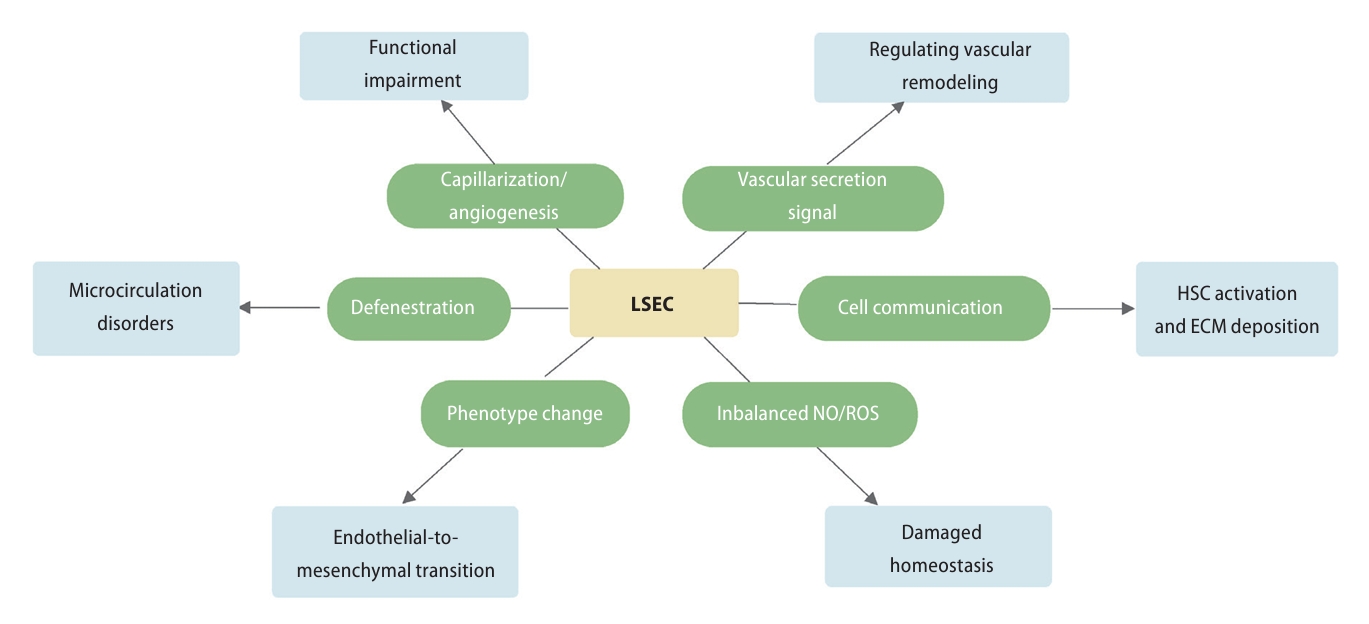
Multiple pathways by which LSECs contribute to the progression of liver fibrosis. During the process of liver fibrosis, LSECs was dedifferentiated, which was characterized as capillarization, defenestration, and phenotype transition. In addition, the intracellular NO/ROS homeostasis and the vascular secretion pathways were remodeling. Further, the impaired communication of LSECs along with several cell types aggravated ECM deposition and the development of liver fibrosis. HSC, hepatic stellate cell; LSEC, liver sinusoidal endothelial cell; NO, nitric oxide; ROS, reactive oxygen species; ECM, extracellular matrix.

Agents regulating liver fibrosis through LSECs
In addition to LSEC capillarization, another significant alteration in liver fibrosis was the reduction or disappearance of LSEC fenestrae, which could reduce liver permeability, thereby hindering the transport of substances within the liver. Given this, restoring the reduction of LSEC fenestrae and maintaining material exchange between blood and Disse space might represent a promising avenue for future liver fibrosis treatments. However, the researches focus on the mechanism of fenestration in LSECs was limited. Among various fenestrated endothelial cells, glomerular endothelial cells (GEnC) exhibit the closest resemblance to LSECs in terms of structural and functional characteristics. The fenestration of GEnC is the key to glomerular filtration barrier, which is similar to that of LSECs as hepatic sinusoid filtration barrier. Given the significant impact of enhancing GEnC fenestrae on the prognosis of chronic kidney disease, it is plausible that the regulation of LSEC fenestrae could similarly contribute to improving liver fibrosis. Mechanistically, it is reported that Eps15 homology domain-containing protein 3 (EHD3) could regulate GEnC fenestrae through endocytosis recirculation of VEGF or interacting with cytoskeleton [113]. Notably, the cytoskeleton remodeling is also closely related to the formation of LSEC fenestration. Zapotoczny et al. [114] have shown that the destabilization of the actin-spectrin scaffold induced by damide and iodoacetic acid (IAA) in LSECs was highly involved in the formation of fenestrations, indicating that spectrin could potentially serve as a therapeutic target for modulating LSEC permeability. Based on these findings, it is crucial to conduct further studies to clarify the pivotal role of the cytoskeleton in LSEC fenestration. Such investigations hold promise as an effective strategy for ameliorating liver fibrosis by governing the LSEC permeability.
We have summarized in this review that when LSECs undergoes EndMT, it currently facilitates the development of liver fibrosis, which is attributed by the loss of original cellular phenotypes and characteristics features in LSECs as well as promoting ECM deposition, one of the main characteristics of liver fibrosis, collectively resulting in abnormal changes in liver structure and function to aggravate liver fibrosis [62]. Yet, limited information was available regarding the instigator of fibrosis associated with EndMT. During the process of liver fibrosis, there is a significant increase in the production of ROS, which exceeds the clearance capacity of cells and leads to an increase in oxidative stress within cells. The surplus ROS in LSECs fosters the EndMT of LSECs, which might be considered as an essential factor to control the EndMT process.
The complex vascular networks in liver play divergent roles in the process of hepatic fibrosis. The increase number of portal vessels alleviates liver fibrosis, while the augmentation of hepatic sinusoidal capillaries and central vessels can aggravate hepatic fibrosis. At present, a considerable number of studies have reported the efficacy of anti-angiogenic drugs in the treatment of hepatic fibrosis, yet the results have shown variability and inconsistency among different investigations [37,38,45-49,56]. One contributing factor to the complicated efficacy may be the underestimation of the microvascular network induced by LSECs. Hence, a more holistic approach to liver fibrosis treatment requires the incorporation of various signaling pathways that regulate endothelial function. This review summarizes the therapeutic effects of different pharmacological interventions on liver fibrosis by inhibiting the expression of angiogenesis-related factors, protecting or restoring LSEC fenestrae, maintaining the LSEC differentiation phenotypes, regulating LSEC vascular secretory signals and affecting communication between LSECs and HSCs. While these agents have shown promising effectiveness in cellular and animal studies, they have not yet progressed to the clinical research phase and still cannot achieve accurate targeting of LSECs. Therefore, exploring therapeutic remedies or administration routes that specifically target LSECs may become a new approach for anti-fibrosis therapy strategy in the future. It is worth noting that the combination administration of adeno-associated viral vector serotype 9 (AAV9)-LECT2-short hairpin RNA (shRNA) specifically targeting LSECs, along with bevacizumab targeting angiogenesis, showed a positive antifibrotic effect and fewer side effects [115]. This indicates that inhibiting the microvascular network initiated by LSECs and other intrahepatic vascular networks concurrently could lead to increased efficacy. Similarly, since hyaluronic acid (HA) could only be phagocytosed by LSECs, it was reported that HA-coupled liposomes nanoparticle could deliver interferon regulatory factor 1 into LSECs in vivo, representing a novel and specific delivery approach [91]. Notably, oncoprotein-induced transcript 3 (Oit3) was considered as a promising hallmark gene for targeting LSEC. Additionally, a liposomal system could deliver compound to specific cells via modified the liposome with specific peptide [116]. Thus, we suggest design a Oit3-targeted peptide combined with liposomal system could achieve the delivery of compound to LSECs, providing a novel direction for the intervention of liver fibrosis.
In addition, another reason for the inefficacy of antiangiogenic drugs may be that regulating a single type of vascular endothelial cell is not enough to fully regulate multiple vascular networks for the treatment of liver fibrosis. Xu et al. [54] reported a new viewpoint concerning capillarization and angiogenesis. Usually, when discussing hepatic angiogenesis, estimating sinusoidal capillarization solely based on counting positive vascular markers (such as CD31) may lead to an inaccurate assessment of capillary formation, as it inappropriately includes portal vein angiogenesis. This means that the inaccurate inference of ‘increased angiogenesis’ might result from combining the counting of positive CD31 markers from portal vessels and sinusoidal capillaries. It is worth mentioning that they did not define sinusoid capillarization as angiogenesis but vascular remodeling, while capillarization and angiogenesis should be regarded as two relatively independent functional processes.
Notes
Authors’ contribution
Xiaojiaoyang Li: Conceptualization, Writing-Review & Editing. Jiaorong Qu: Writing-Review & Editing, Investigation, Visualization. Le Wang: Investigation, Writing-Original Draft. Yufei Li: Investigation.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
Acknowledgements
This work was supported by National Natural Science Foundation of China (Grant NO. 82274186 to XL); National Key Research and Development Program on Modernization of Traditional Chinese Medicine (Grant NO. 2022YFC3502100 to XL); National High-Level Talents Special Support Program to XL.
Abbreviations
A-FABP
adipocyte fatty acid binding protein
AKAP12
A-kinase anchoring protein 12
Ang2
angiopoietin 2
AT1R
angiotensin II type 1 receptor
BMP9
bone morphogenetic protein 9
BRG1
brahma-related gene 1
CCL2
C-C motif chemokine ligand 2
CO
carbon monoxide
CXCR
C-X-C chemokine receptor
DLL4
delta-like ligand 4
ECM
extracellular matrix
EndMT
endothelial-mesenchymal transition
eNOS
endothelial nitric oxide synthase
ERK
extracellular-signal-regulated kinase
FGFR1
fibroblast growth factor receptor 1
GATA4
GATA binding protein 4
HGF
hepatocyte growth factor
Hh
hedgehog
HIF-1α
hypoxia inducible factor-1α
HMGB1
high-mobility group box 1
HSCs
hepatic stellate cells
ICAM1
intercellular adhesion molecule 1
JAM
junctional adhesion molecules
KLF5
Krüppel-like factor 5
LDH-A
lactate dehydrogenase-A
LECT2
leukocyte cell-derived chemotaxin 2
LSECs
liver sinusoidal endothelial cells
LXRs
liver X receptors
MASLD
metabolic dysfunction-related fatty liver disease
NADPH
nicotinamide adenine dinucleotide phosphate
NASH
nonalcoholic steatohepatitis
NF-κB
nuclear factor kappa B
NO
nitric oxide
NOX4
NADPH oxidase 4
NRP-1
Neuropilin-1
PDGF
platelet-derived growth factor
PDGFR-β
platelet-derived growth factor-receptor β
PI3K
phosphatidylinositol 3'-kinase
PLA2s
phospholipase A2s
ROS
reactive oxygen species
SIPR2
sphingosine-1-phosphate receptor 2
SPHK1
sphingosine kinase 1
STAT3
signal transducer and activator of transcription 3
TGF
transforming growth factor
TMP
tetramethylpyrazine
TNF-α
tumor necrosis factor-alpha
VCAM1
vascular cell adhesion protein 1
VEGF
vascular endothelial growth factor
VEGFR
vascular endothelial growth factor receptor
YAP
Yes-associated protein