Graphical Abstract
INTRODUCTION
Despite the availability of preventive vaccines, hepatitis B virus (HBV) infection remains a global health concern, affecting over 257 million individuals worldwide. Chronic hepatitis B (CHB) is associated with serious complications, such as liver failure, cirrhosis, and hepatocellular carcinoma (HCC) [1]. HBV contains partial double-stranded DNA that is converted into the covalently closed circular DNA (cccDNA) minichromosome, which acts as a transcription template to produce viral RNAs. HBV reverse transcriptase/ polymerase synthesizes viral DNA from pre-genomic RNA (pgRNA) inside newly formed nucleocapsids. Although the life cycle of HBV and innate immune response to HBV infection have been extensively studied, the network of host factors and signaling pathways that could potentially exert inhibitory effects on HBV replication is not fully understood [2].
The 3.2 kb HBV genome encodes four proteins: polymerase (P), surface (S; pre-S1, pre-S2, and S), precore/ core (C), and X (HBx). HBx is a key factor that promotes the transcription and replication of HBV and is known to modulate immune avoidance by regulating host antiviral signaling pathways and molecules. HBx consists of 154 amino acids, including a transactivation domain, a nuclear translocation domain, and a negative regulatory domain. The mechanism by which HBV escapes the immune system remains unclear [3,4].
Various cytokines have been reported to inhibit HBV transcription and replication via diverse mechanisms both in vitro and in vivo [5]. For instance, tumor necrosis factor-alpha (TNF-α) and interferon gamma (IFN-γ) are secreted by HBV-specific CD8+ T cells. TNF-α reduces HBV transcription and capsid stability, whereas IFN-γ has been reported to eliminate pgRNA-containing capsids in a HBV-infected mouse model [6,7]. In addition, pegylated-IFN-α (Peg-IFN-α) is currently used to treat HBV infections along with nucleos(t)ide analogs (NAs) [8,9]. Although NAs target viral reverse transcriptase to inhibit viral replication, they cannot completely eradicate CHB due to the persistence of HBV cccDNA within infected hepatocytes [10]. Moreover, a major drawback of utilizing IFN-α as a therapeutic agent is its association with multiple side effects [11]. Therefore, currently available therapies could be combined with targeted suppression of viral RNA and cccDNA to achieve more robust therapeutic strategies. Accordingly, ongoing efforts to exploit the interplay between the virus and host factors, with a particular focus on uncovering anti-HBV host factors, are essential for obtaining a novel strategy for a functional cure.
The major histocompatibility complex (MHC) class II transactivator (CIITA) is a regulatory transcription factor, whose expression is induced by IFN-γ [7]. It plays a central role in stimulating the immune response against infections by elevating the expression of class II MHC molecules within antigen-presenting cells (APCs) [12,13]. Interestingly, CIITA may be involved in antiviral defense through the intracellular regulation of other host factors required for virus replication [14-16]. In addition, CIITA induction confers cellular resistance to Ebola virus and severe acute respiratory syndrome (SARS)-like coronaviruses. Moreover, there is evidence of a relationship between HBV and CIITA, and single-nucleotide polymorphisms (SNPs) in CIITA have been shown to be associated with CHB infection [17].
Here, we investigated the inhibitory function of CIITA against HBV and explored its mechanism of action. We found that CIITA has an IFN-mediated anti-HBV activity, that inhibited the transcription of HBV by suppressing the main HBV enhancers/promoters. In addition, it reduced the hepatocyte nuclear factor (HNF)4α levels through extracellular signal-regulated kinase (ERK1/2) pathway which further restricts viral transcription and replication in hepatocytes. CIITA overexpression impaired HBV DNA replication in both patient-derived primary human hepatocytes (PHH) and in vivo mouse systems. Intriguingly, HBx interacted with the CIITA protein, resulting in resistance to HBV inhibition by CIITA. Hence, our investigation has unveiled a novel transcription regulator with an anti-HBV function that can be explored further for potential therapeutic strategies to combat HBV infection.
MATERIALS AND METHODS
Cell culture
The human HCC cell line HepG2 (American Type Culture Collection, ATCC no. HB-8065), Huh7 (Korean Cell Line Bank, KCLB), and HepG2-NTCP cells [18] were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Welgene, Gyeongsan, Korea) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin streptomycin (PS; Gibco, Grand Island, NY, USA) at 37°C in a 5% CO2 humidified incubator. HepAD38 was maintained by adding 0.3 μg/mL tetracycline to the same media as HepG2.
Isolation of PHHs from patient tissues
PHHs were obtained via therapeutic hepatectomy in donors who tested negative for hepatitis A virus (HAV), HBV, hepatitis C virus (HCV), or hepatitis D virus (HDV). Informed consent was obtained from both patients (66-yearold male and 89-year-old male) prior to surgery, and this study was approved by the Institutional Review Board of Korea University Hospital (IRB no. ED10287). PHHs were isolated using a two-step collagenase perfusion method, as described previously [19]. Isolated PHHs were seeded on collagen-coated plates (Corning, Tewksbury, MA, USA) in William’s E medium (Gibco) containing cell maintenance supplements (CM4000; Gibco), 2% FBS, and 1% penicillin/ streptomycin.
Plasmids and siRNA transfection
HBV 1.2-mer wild type (WT) replicon (HBV 1.2 (+)) and HBx-null 1.2-mer (HBV 1.2 (-)) have been described previously [20]. The pcDNA3-myc-CIITA (P#808, addgene) plasmid was purchased from Addgene (P#808). The siRNA Negative Control (AccuTarget™ Negative Control siRNA [BioRP, 20nmole] SN-1003) and the siCIITA (Sense: GGAGCUUCUUAACAGCGAU Antisense: AUCGCUGUUAAGAAGCUCC) were obtained from Bioneer (Daejeon, Korea). Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA, USA) was used for plasmid transfection, and Lipofectamine RNA/iMAx (Thermo Fisher Scientific) was used for siRNA transfection. Transfection experiments were performed according to the manufacturer’s instructions.
HBV infection
The HBV inoculum was prepared from the culture supernatant of HepAD38 cells according to a previously described protocol [21]. HepG2-NTCP or PHH cells were seeded onto 6-well plates coated with collagen I (Gibco) and infected with the HBV inoculum in DMEM supplemented with 4% PEG 8000 (Sigma, Darmstadt, Germany) overnight. Thereafter, the cells were washed three times with PBS, maintained in DMEM containing 2.5% DMSO, and harvested at one-week post-infection.
Southern and Northern blot analysis
HBV DNA replication was analyzed using Southern blotting as described previously, with minor modifications [22]. Three days after transfection, cells were harvested and lysed with HEPES NP-40 lysis buffer. After centrifugation, the supernatant was separated and treated with DNase I at 37°C for 20 min to remove the transfected plasmid. The samples were incubated for at least 1 h on ice with a polyethylene glycol solution (PEG 8000; Merck, Darmstadt, Germany) to precipitate the HBV core particles. Proteinase K (Merck) in SDS solution was added to the samples to disrupt the capsid structure. HBV capsid-associated DNA was purified using phenol-chloroform-isoamyl alcohol (25:24:1) (Merck) before precipitation in 100% ethanol and 3 M sodium acetate. Total DNA was separated using electrophoresis on a 1% agarose gel at 90 V for 3 h and transferred to a positively charged nylon membrane (Merck). Hybridization with a digoxigenin (DIG)-labeled DNA probe was performed, and Southern blot signals were detected using a DIG Nucleic Acid Detection Kit (Roche, Mannheim, Germany). Signals were detected using ImageQuant 800 (Amersham, Buckinghamshire, UK). RNA was extracted using TRIzol reagent (Sigma) according to the manufacturer’s protocol, and 20 μg of total RNA was separated on a 1.5% formaldehyde-agarose gel in 1X MOPS buffer (Biosesang, Seongnam, Korea). 28S and 18S rRNAs were used as controls. After electrophoresis, the protocol was performed in the same manner as that for the Southern blot analysis.
cccDNA extraction
Hirt DNA was extracted as described previously [23]. Briefly, the cells were lysed using Hirt lysis buffer (50 mM Tris–HCl [pH 7.5], 10 mM EDTA, 150 mM NaCl, and 1% SDS). After high salt precipitation using 2.5 M KCl, the DNA was subjected to phenol-chloroform extraction followed by isopropanol precipitation. Subsequently, cccDNA was purified by ethanol precipitation and resuspended in Tris-EDTA (TE) buffer. To test the authenticity of cccDNA, a standard procedure involving boiling at 88°C and subsequent EcoRI digestion was applied [24]. Phosphonoformic acid (PFA) was added to the cells to block cccDNA formation [25] and then was removed after four days. The prepared cccDNA was quantified using Southern blotting.
Western blotting
To analyze the protein levels, cells were harvested using RIPA buffer containing a protease inhibitor cocktail (Merck) for 30 min on ice. The supernatant was transferred to a new e-tube after centrifugation at 13,000 RPM for 20 min. The samples were mixed with Laemmli sample buffer (BioRad, Hercules, CA, USA) and boiled at 95°C for 5 min, followed by chilling on ice. Then, samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for 2 h at 80 V and transferred onto a polyvinylidene fluoride (PVDF) membrane using the Trans-blot Turbo RTA PVDF Transfer Kit (Bio-Rad). Membranes were washed with TBS-T (20 mM Tris, 150 mM NaCl, and 0.1% Tween 20). After blocking with 5% skim milk, membranes were shaken overnight at 4°C with a primary antibody solution containing 3% BSA. The membranes were washed three times with TBS-T and incubated with a secondary antibody solution containing 3% skim milk at room temperature for 1 h. Membranes were visualized by ECL (Abclon, Seoul, Korea) using ImageQuant 800 (Amersham). The primary antibodies used in this study are listed in Supplementary Table 1.
Enzyme-linked immunosorbent assay (ELISA)
The cell supernatants were collected to quantify the secretion levels of HBeAg and HBsAg after dilution with PBS. The secreted HBV antigen levels were measured at an optical density (OD) of 450 nm using an ELISA kit (Wantai Pharm Inc., Beijing, China) and a spectrophotometer (SpectraMAX Plus 384).
Luciferase reporter assay
Two days after transfection, cells were lysed, and luciferase activity was measured using the Luciferase Assay System (Promega, Madison, WI, USA). β-galactosidase activity was determined using a β-galactosidase enzyme assay system (Promega) to confirm the transfection yield.
Co-immunoprecipitation (Co-IP)
Cells were treated with 20 μM MG132 for 5 h before harvesting. Initially, harvested cells were lysed using Pierce™ IP Lysis Buffer (Thermo Fisher Scientific, 87787) containing protease inhibitor cocktail (Merck). Then, 20 μL lysate was transferred to new micro-tube, mixed with Laemmli sample buffer (Bio-Rad) and retained as the input. For IP, the supernatant was incubated with a primary antibody at 4°C overnight. Protein A-agarose beads (Merck) were added, and the samples were rotated for 4 h. After centrifugation at 3,000 rpm for 1 min, the supernatant was discarded and the beads were washed with PBS three times. The immune complexes were separated using SDS-PAGE, and protein signals were examined using Western blotting.
Quantitative real-time PCR
Total cellular RNA was isolated, and CIITA, STAT-A, HNF4α, HNF1α, HNF3β, C/EBPα, GAPDH, and HBV DNA expression were analyzed using real-time PCR. Reverse transcription was performed using 2 μg of total RNA and a high-capacity RNA-to-cDNA kit (Thermo Fisher Scientific). Quantitative real-time PCR was performed in the QuantStudioTM 5 Real-Time PCR System using SYBR Green PCR Master Mix (Applied Biosystems). The results were expressed as the fold difference relative to the calibrator, determined using the ΔΔCT method. The specific primer sequences used for real-time PCR are listed in Supplementary Table 2.
Hydrodynamic injection in mice
Six-week-old male C57BL/6 mice were hydrodynamically injected with HBV 1.2 (+) and Myc-CIITA plasmids at a volume equivalent to 10% of the mouse body weight. All animal experiments were approved by the Animal Care Committee of Sungkyunkwan University. Four days after injection, the mice were sacrificed, and the liver tissues and serum were isolated for Southern blotting and ELISA, respectively.
Confocal microscopy
To evaluate the co-expression levels of HBV 1.2-mer and FAM-negative control siRNA (Bioneer), confocal microscopy analysis was performed. Huh7 cells grown on cover slides were incubated overnight at 4°C with HBcAg primary antibody (1:300) containing 3% BSA in PBS. Cells were washed with PBS and incubated with secondary antibody conjugated with Alexa 568 to detect red signal. The fluorescence signals were visualized using a TCS SP8 HyVolution confocal laser scanning microscope (Leica Microsystems CMS GmbH). The images were analyzed using LAS X software (Leica Microsystems).
RESULTS
CIITA suppresses HBV replication in HCC cell lines and mediates the anti-HBV activity of IFN-γ
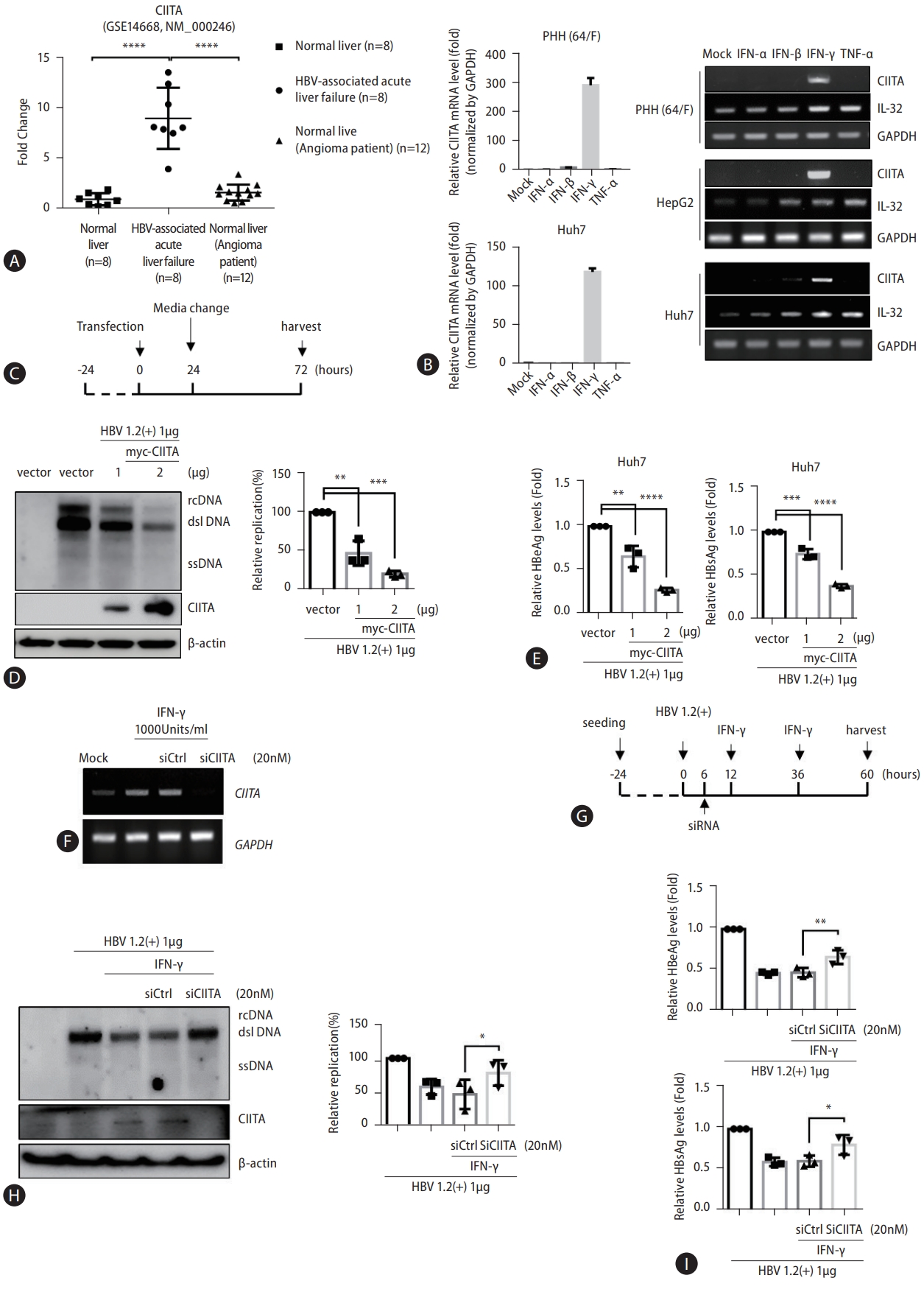
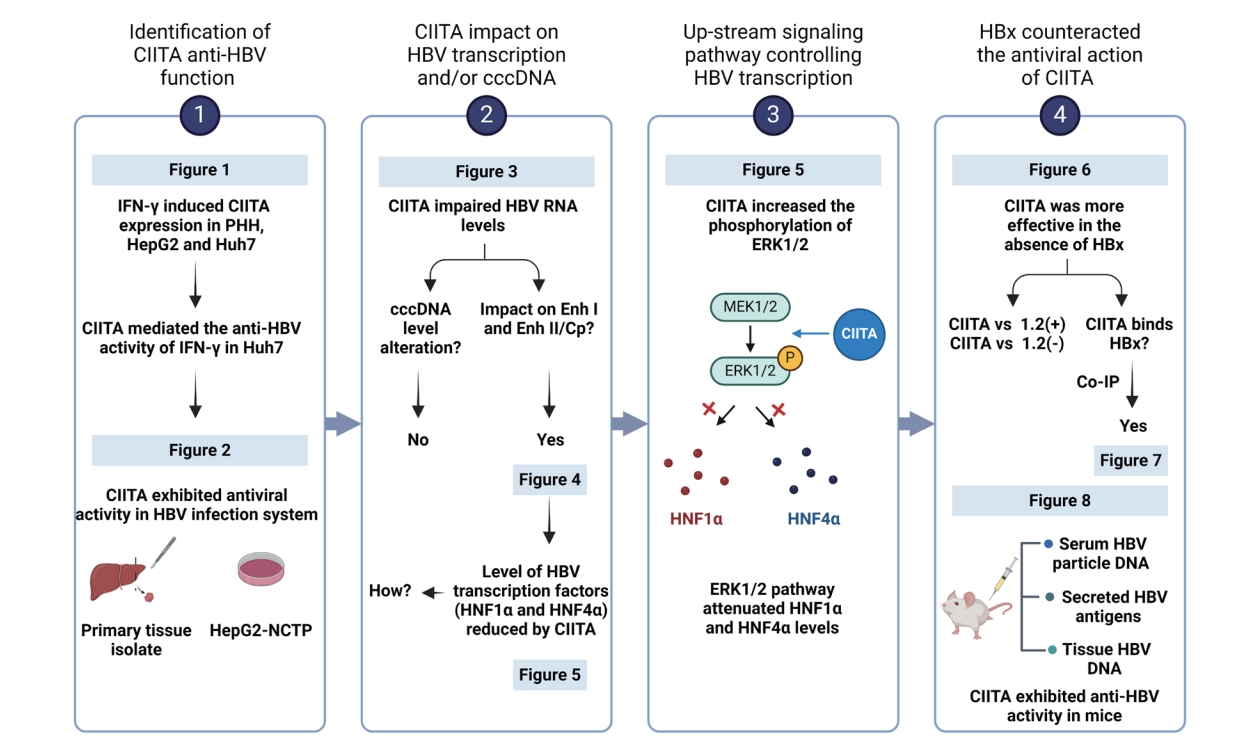
Initially, through new analysis of previously obtained gene expression profiles, we identified a correlation between HBV infection and CIITA gene expression. The specimens used for this analysis were obtained from HBV-negative normal liver donors (or patients who underwent liver resection for hemangioma) and donors with HBV-associated acute liver failure [26]. While CIITA levels remained unchanged in the normal liver or non-HBV-related hemangioma patients, we observed a significant increase (between 5- to 15-fold increase) in the gene expression of CIITA in eight patients with acute liver failure caused by HBV (Fig. 1A). According to previous reports, CIITA expression is induced by IFN-γ, and IFN-γ, along with TNF-α, is associated with HBV viral clearance [27]. Therefore, to determine whether CIITA expression is actually induced by IFN-γ in hepatocytes, HepG2, Huh7, and PHH cells were treated with different cytokines; CIITA induction levels were measured. Semi-quantitative RT-PCR results showed that CIITA expression was induced by IFN-γ in the two HCC cell lines as well as PHH (Fig. 1B-right). Interleukin-32γ (IL32) gene expression levels were measured as the positive control [28]. In the tissues isolated from a PHH donor and Huh7 cells, CIITA mRNA levels were substantially increased, as demonstrated by real-time PCR (Fig. 1B-left). To explore the anti-HBV activity of CIITA, Huh7 cells were co-transfected with the HBV 1.2 (+) replicon and the myc-CIITA plasmid. The experimental scheme is shown in Figure 1C. As shown in Figure 1D, CIITA reduced HBV replication in a dose-dependent manner as determined by Southern blotting. Similarly, the ELISA results showed that the levels of HBeAg and HBsAg secreted in the cell supernatant significantly decreased (Fig. 1E).
After evaluating the function of the siRNA by measuring CIITA RNA levels (Fig. 1F), we silenced the expression of CIITA using 20 nM of siCIITA (Fig. 1F) and treated the Huh7 cells with IFN-γ, as shown in the scheme in (Fig. 1G). The co-transfection efficiency was examined by confocal microscopy (Supplementary Fig. 1). FN-γ-mediated reduction of HBV replication was reversed by CIITA depletion (Fig. 1H). Moreover, HBeAg and HBsAg levels in the cell supernatant were similarly restored (Fig. 1I). Collectively, these findings suggest that CIITA inhibits HBV replication and partially mediates IFN-γ-induced anti-HBV activity in HCC cells.
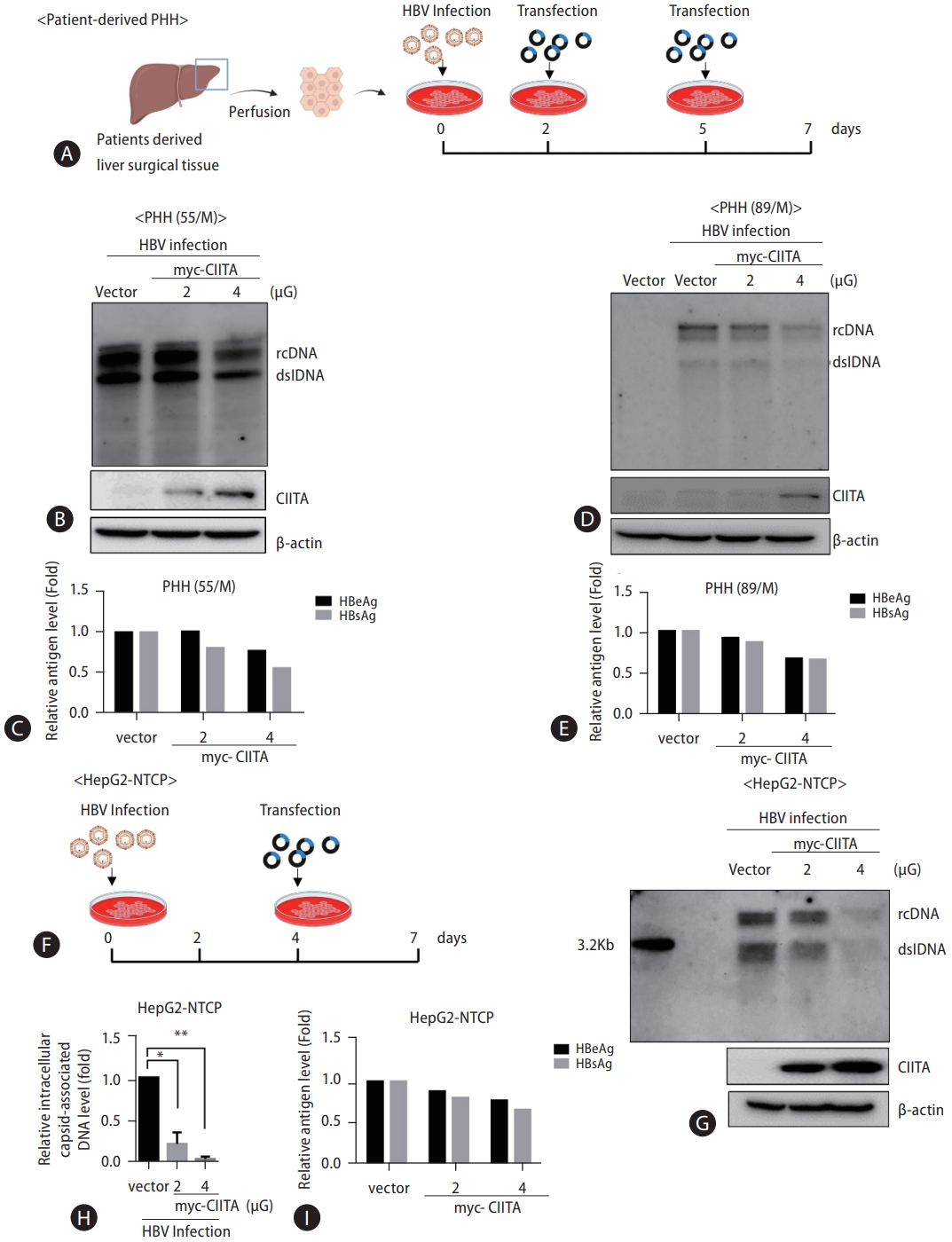
CIITA exhibits antiviral activity in the HBV infection system
Next, we validated the effects of CIITA in a biologically relevant HBV infection system by isolating PHHs from two liver tissue donors (Fig. 2A). In both donor samples, CIITA diminished HBV DNA replication levels in a dose-dependent manner, consistent with the decline in HBsAg and HBeAg levels (Fig. 2B–E). A similar experiment was conducted using HepG2-NTCP cells, which are a well-established infection model, to explore the anti-HBV effects of CIITA. In line with the results in PHHs, the intracellular capsid-associated HBV DNA and secreted antigen levels were reduced in a concentration-dependent manner (Fig. 2F–I). Collectively, these findings indicate that CIITA displays antiviral properties against HBV in an actual infection system.
CIITA impairs HBV at the transcriptional level but does not exert regulatory control over cccDNA levels
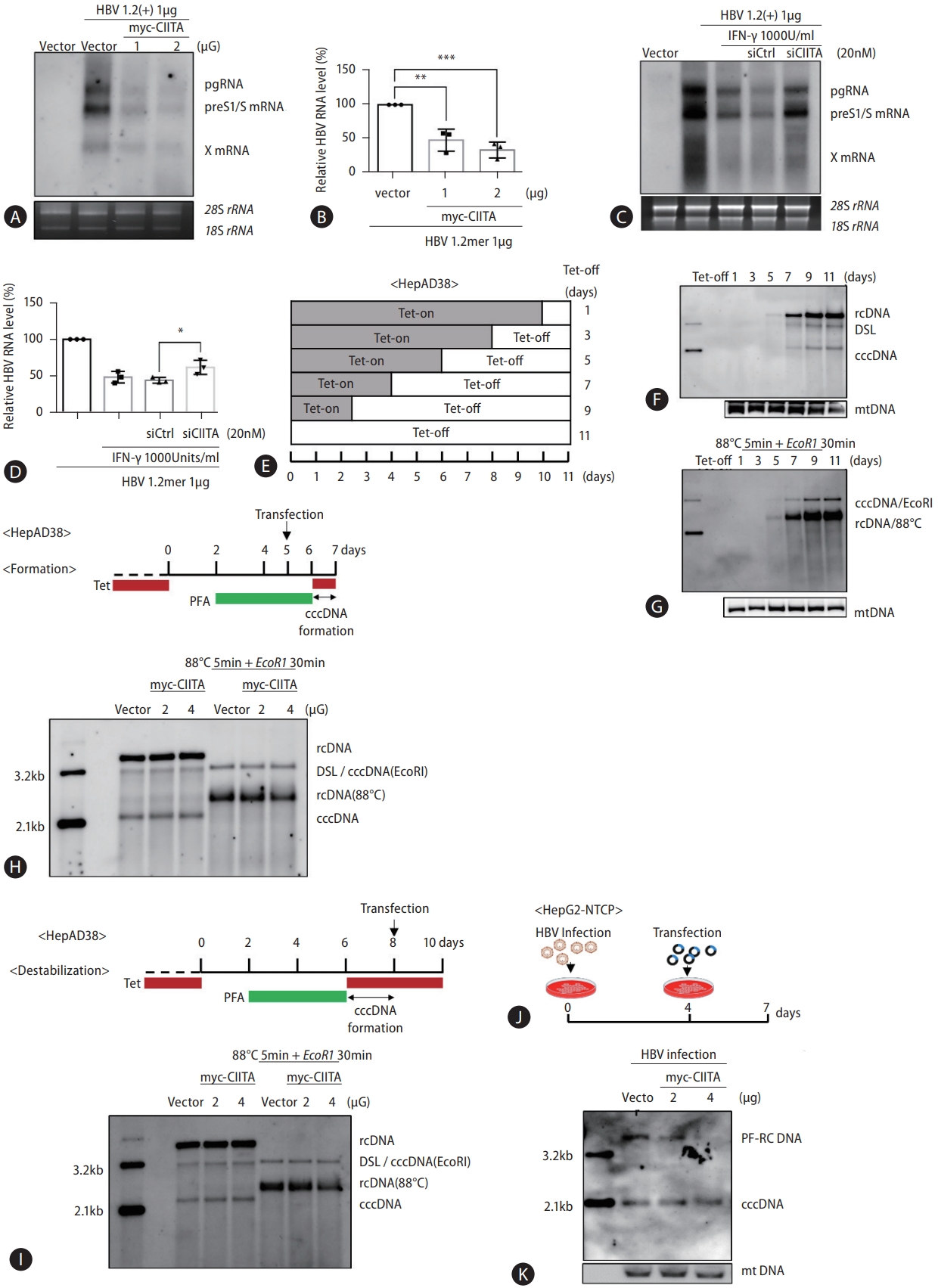
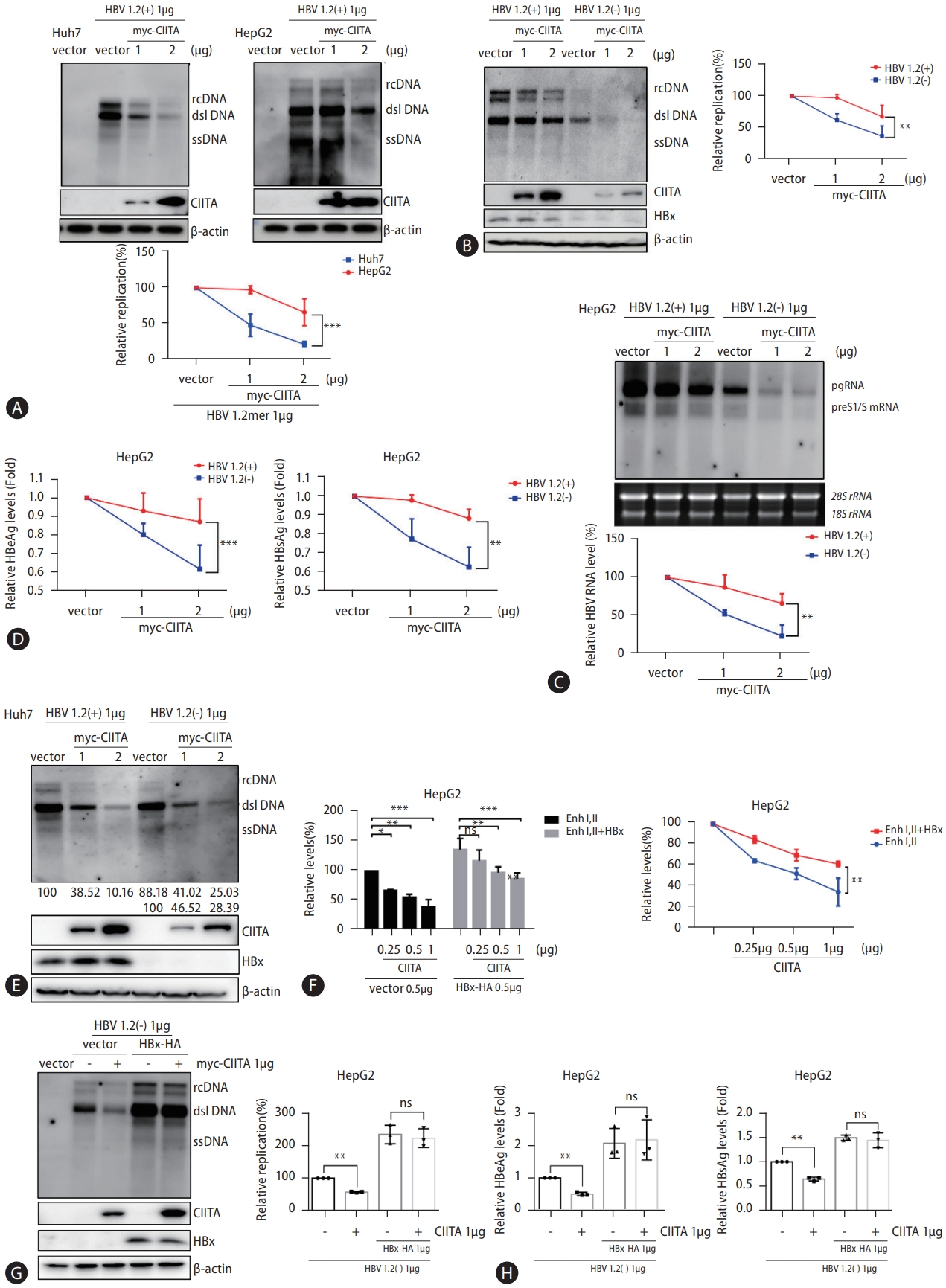
Considering that the conventional function of CIITA is as a major regulator of MHC class II transcription [29], we wondered whether CIITA inhibited the transcriptional activity of HBV. The experimental flowchart of the mechanistic study is summarized in Supplementary Figure 2. Following the ectopic expression or knockdown of CIITA, HBV transcripts were examined using Northern blotting. CIITA overexpression decreased pgRNA, preS1/S mRNA, and X mRNA levels by more than 50% (Fig. 3A, B). In addition, after silencing CIITA, IFN-γ failed to efficiently reduce HBV RNA levels (Fig. 3C, D), suggesting that CIITA partially mediates the anti-HBV activity of IFN-γ. These results imply that CIITA induces HBV resistance by affecting viral gene expression and is involved in IFN-γ-mediated anti-HBV activity.
Since HBV RNA levels were significantly decreased in cells transfected with CIITA, we sought to determine whether the impact of CIITA extended to cccDNA alterations. After investigating the kinetics of cccDNA in HepAD38 cells (Fig. 3E–G), a suitable strategy for the transfection of the CIITA plasmid was employed (Fig. 3H and I, upper panel). The robust formation of cccDNA was detected at 7 d and reached a plateau at 9 d after Tet-off (Fig. 3F, G). Therefore, we harvested the cells 7 and 10 days after Tet-off to determine the impact of CIITA on cccDNA formation and destabilization. Furthermore, PFA was used to arrest HBV DNA synthesis, which allows for the time-dependent formation of cccDNA [25]. Intriguingly, CIITA had no noticeable effect on cccDNA formation or destabilization (Fig. 3H and I bottom). We confirmed these results in an HBV infection model using HepG2-NTCP cells (Fig. 3J, K). Nonetheless, the levels of HBV cccDNA remained constant despite transient transfection with increasing doses of CIITA plasmid. These observations imply that CIITA may regulate HBV transcription rather than directly affect cccDNA abundance.
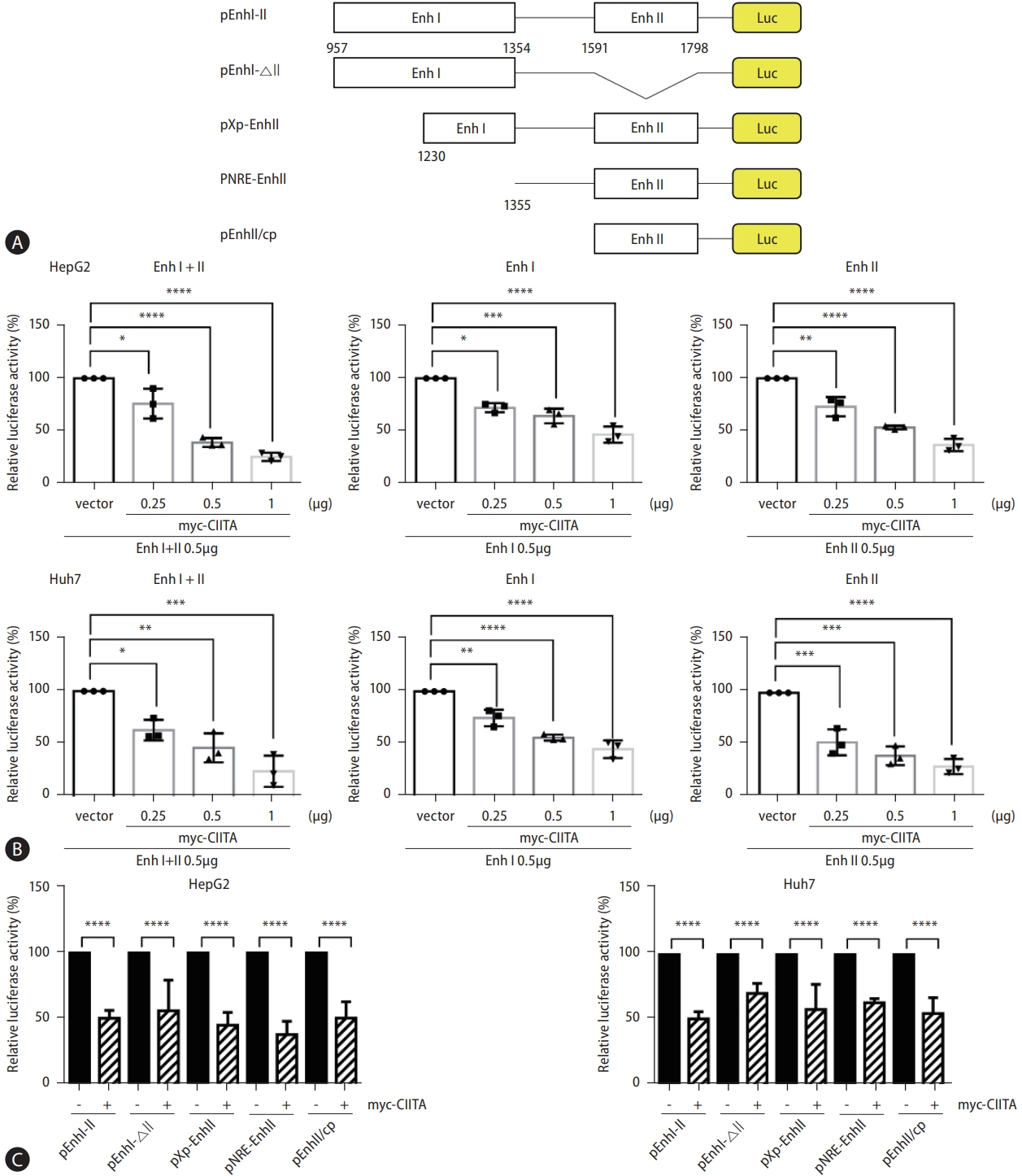
CIITA suppresses activity of HBV enhancers and promoters
HBV transcription is regulated by enhancers I (Enh I) and II (Enh II), the latter of which overlaps with the core promoter (Cp). Since CIITA exhibited pronounced inhibition of HBV transcription without affecting cccDNA, we examined the possible impact of CIITA on the HBV enhancer and promoter regions using a luciferase reporter assay (Fig. 4A) [28]. The results demonstrated that the concentration-dependent overexpression of CIITA caused a notable reduction in the activity of each enhancer (Fig. 4B). We reasoned that the classical function of CIITA is to regulate transcription by binding to the promoter of MHC class II genes; therefore, we determined whether CIITA directly affects specific regions of the HBV enhancer and promoter via deletion mutants. A downward trend was detected in the activity of all enhancer sites in both HepG2 and Huh7 cells transfected with the CIITA plasmid (Fig. 4C), highlighting the ability of CIITA to simultaneously suppress HBV Enh I and Enh II/ Cp.
CIITA reduces the expression of major hepatocyte nuclear factors through the ERK1/2 pathway
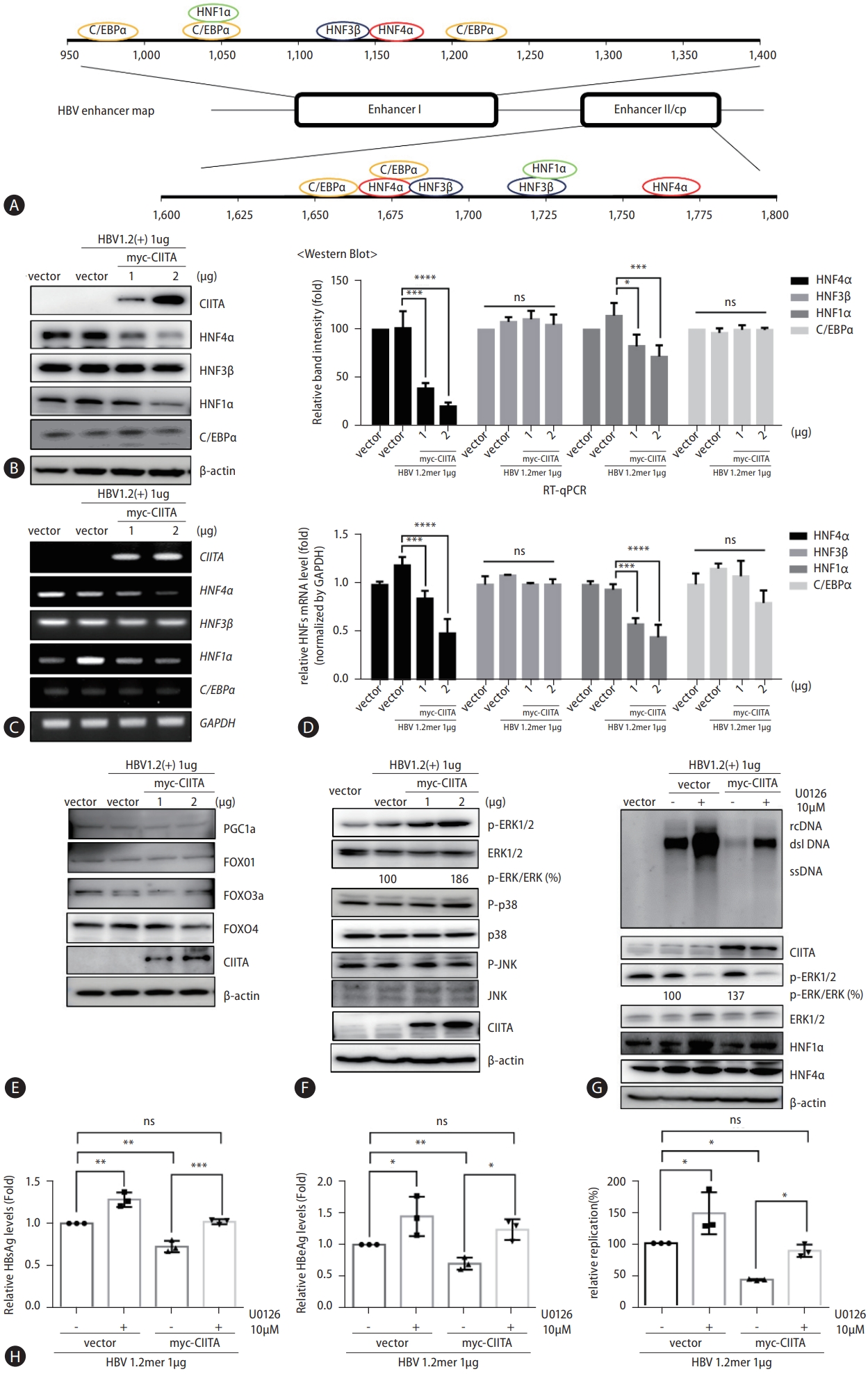
CIITA exhibited a broad inhibitory effect on the activity of most HBV enhancers (Fig. 4). Transcriptional regulation of HBV involves the engagement of various transcriptional regulatory factors within hepatocytes [30]. We hypothesized that, as a master regulator of transcription, CIITA might exert control over the expression of essential liver-enriched transcription factors involved in the transcription of both Enh I and Enh II of HBV, including C/EBPα, HNF1α, HNF3β, and HNF4α (Fig. 5A). Our findings revealed a reduction in both the protein (Fig. 5B) and mRNA levels (Fig. 5C and D) of HNF1α and HNF4α in response to CIITA. However, the levels of HNF3β and C/EBPα did not significantly differ (Fig. 5B–D). These results indicated that CIITA exerts its anti-HBV effects by downregulating transcription factors that are essential components of the virus life cycle.
HNFs are regulated by diverse regulatory proteins and signaling pathways, including PGC1a, the FOXO family, and mitogen-activated protein kinase (MAPK) [31,32]. Consequently, to explore the mechanism by which CIITA downregulates the expression of HNF4α and HNF1α, we examined the activation of different transcription factors known to regulate HNF4α expression. Western blotting results showed that ectopic CIITA protein expression had no significant impact on PGC1a, FOXO1, FOXO3a, and FOXO4 protein levels in Huh7 cells (Fig. 5E). Therefore, we investigated the activity of MAPK signaling pathway components using a similar experiment. CIITA activated ERK1/2 by significantly increasing its phosphorylation. However, increasing dose of CIITA did not affect the phosphorylation levels of p38 and Jun N-terminal kinase (JNK) phosphorylation levels (Fig. 5F). To further determine whether the main mechanism through which CIITA suppresses HBV is by activating the ERK pathway, we treated cells with U0126, a highly selective inhibitor of the ERK pathway. As expected, phosphorylation of ERK1/2 (p-ERK1/2) was attenuated in the cells treated with U0126 (Fig. 5G, bottom). Interestingly, the antiviral effects of CIITA were largely nullified in the presence of U0126, whereas ERK1/2 levels remained unchanged (Fig. 5G and H). The reduction of HNF1α and HNF4α levels by CIITA was also restored following U0126 treatment. These observations indicate that the mechanism underlying CIITA-mediated downregulation of HBV transcription involves the induction of the ERK1/2 pathway, which eventually leads to a remarkable reduction in the levels of HNF1α and HNF4α.
HBx protein counteracts the antiviral function of CIITA
To further validate the effect of CIITA on HBV replication in various HCC cell lines, we conducted parallel experiments using HepG2 cells. Although the inhibitory effect of CIITA on HBV enhancers was consistent in HepG2 and Huh7 cells, the effect of CIITA on HBV replication was less pronounced in HepG2 cells (Fig. 6A). HBx protein expression was lower in Huh7 cells than in HepG2 cells. Although this may be predominantly dependent on cell line characteristics, the fact that HBx has been identified as a pivotal orchestrator of immune evasion mechanisms led us to wonder whether HBx counteracts CIITA antiviral activity. We found that the HBV DNA, RNA, and secreted protein levels decreased less drastically in HepG2 cells following CIITA treatment. Moreover, to achieve a significant reduction in HBV transcription and replication, HepG2 cells required more than double the amount of CIITA protein than Huh7 cells (1 vs. 2 μg in Huh7 and HepG2 cells, respectively) (Fig. 6A). To further test the effect of HBx on the activity of CIITA, HepG2 cells were co-transfected with wild-type HBV 1.2 (+) or HBx minus HBV 1.2 (-) plasmids, along with increasing doses of the CIITA construct. Although there was a slight reduction in the levels of HBV DNA, RNA, and secreted antigens in the presence of the HBx-expressing 1.2 (+) plasmid, cells transfected with the HBx-null 1.2 (-) replicon exhibited a more rapid reduction in the levels of HBV components (Fig. 6B–D). A similar experiment was performed using Huh7 cells, which revealed no discernible differences in the cells between the presence or absence of HBx (Fig. 6E).
The influence of CIITA on HBV promoter activity was also mitigated by the HBx protein (Fig. 6F). To confirm that this counteraction was caused by HBx specifically, co-transfection of HBx-HA and Myc-CIITA plasmids was performed. Unsurprisingly, HBV replication levels were dramatically increased following the ectopic expression of HBx (Fig. 6G). Notably, HBx overexpression resulted in disappearance of the antiviral effect of CIITA, as HBV core-associated DNA levels remained the same in the presence of HBx and CIITA (Fig. 6G). Similar results were observed for HBeAg and HBsAg levels, as demonstrated by ELISA (Fig. 6H). Collectively, these findings implied that HBx interfered with the antiviral activity of CIITA.
HBx protein interacts with CIITA
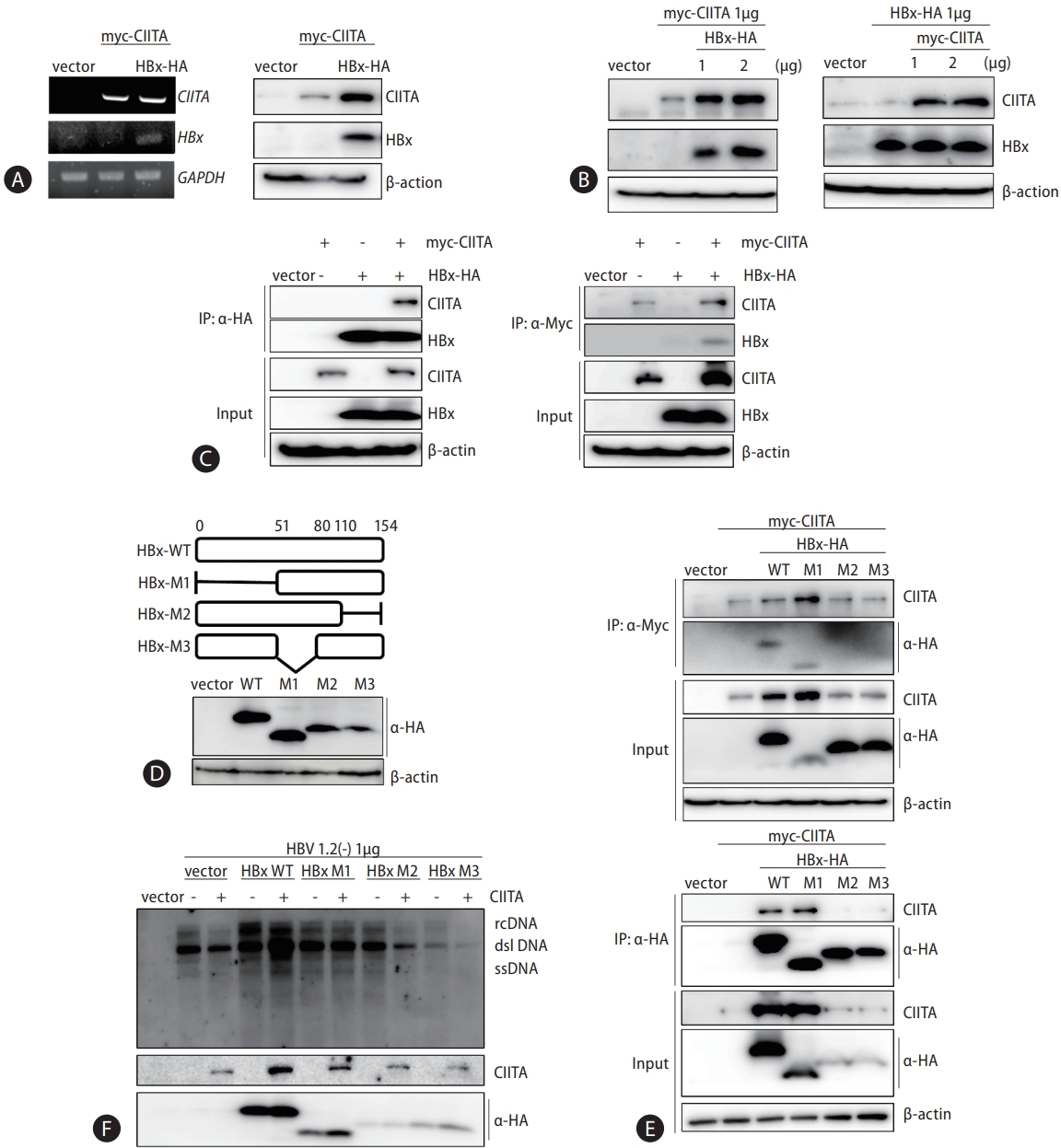
The above data revealed an unexpected increase in CIITA protein levels in the presence of HBx (Fig. 6G). Therefore, we investigated the possible mutual effects of these two proteins by co-transfecting HBx-HA and myc-CIITA plasmids in HepG2 cells. The presence of HBx led to a dose-dependent increase in CIITA protein levels without affecting its mRNA expression (Fig. 7A). However, the HBx protein levels remained unchanged in cells transfected with CIITA (Fig. 7B). To elucidate the interplay between an increase in CIITA protein levels and a reduction in its antiviral effect by HBx, we hypothesized that there might be a direct interaction between the two proteins, which eventually leads to stabilization or deactivation of the CIITA protein.
Accordingly, Co-IP was performed on HepG2 cells expressing myc-CIITA and HBx-HA plasmids, and proteinprotein binding was confirmed using western blotting (Fig. 7C). To determine the critical region of HBx for binding to CIITA, deletion mutants of HBx were used (Fig. 7D). Remarkably, CIITA interacted with WT and mutant 1 (M1) HBx, which corresponded to the observed increase in CIITA protein levels (Fig. 7E, upper panel). Moreover, the CIITA signal was only detectable following IP against WT and M1 HBx (Fig. 7E, bottom panel). To further validate the effect of HBx mutants on HBV replication, plasmids containing HBx WT or mutant variants were introduced into cells along with the HBx-null HBV replicon (HBV 1.2 (-)). Subsequently, we assessed the anti-HBV activity of CIITA using Southern blot analysis. As depicted in Figure 7F, in the presence of WT and M1 HBx, CIITA was incapable of preventing HBV replication. Conversely, M2 and M3 HBx did not interfere with the CIITA function. This indicates that the amino acid sequence 51–154 of HBx bind to CIITA and plays a critical role in inhibiting the anti-HBV activity of CIITA.
CIITA exhibits anti-HBV activity in an in vivo mouse model
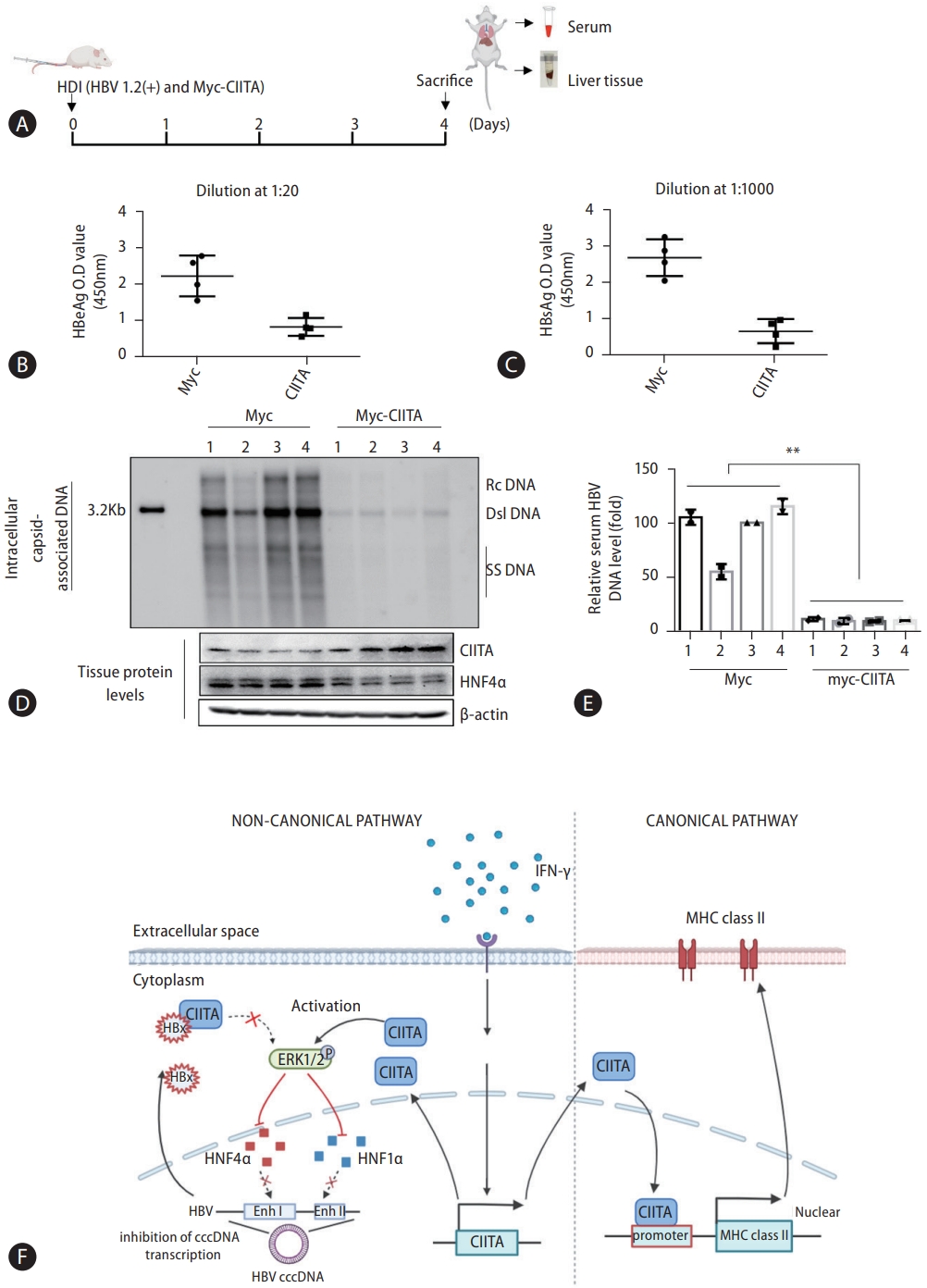
Finally, we examined the in vivo antiviral effect of CIITA using an HBV mouse hydrodynamic injection (HDI) model. Mice were injected with the full HBV genome, to establish a transient HBV transgenic infection state, along with the CIITA plasmid construct. The control group was injected with the HBV construct and the Myc plasmid. Four days after injection, serum samples were collected to measure HBeAg and HBsAg levels and liver tissues were homogenized to quantify HBV DNA levels (Fig. 8A). As depicted in Figure 8B–D, CIITA exerted substantial viral suppression in mice, as indicated by the significantly reduced HBV antigen (Fig. 8B and C) and DNA levels in mouse liver tissues (Fig. 8D, upper panel) as well as serum (Fig. 8E). Furthermore, HNF4α expression levels in liver tissues were determined using western blot analysis (Fig. 8D, bottom panel). Notably, in the mice administered the CIITA plasmid, a significant decrease in HNF4α levels was observed. In summary, these findings demonstrated that CIITA exerts an anti-HBV effect in vivo.
DISCUSSION
Cytokines play an important regulatory role in HBV transcription and replication; however, the precise mechanisms underlying this regulation remain unclear [6,33,34]. Therefore, identifying HBV-associated host factors that are influenced by cytokines can yield valuable insights into antiviral immunity as well as novel therapeutic agents. Notably, IFN-γactivated CIITA, known for its role in MHC class II antigen presentation, has been suggested to have roles beyond its canonical function. Remarkably, a report revealed that CIITA activation confers cellular resistance against the Ebola virus and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [15]. These findings prompted us to explore the potential of CIITA to inhibit HBV gene expression. In this study, we found that CIITA impairs HBV at the transcriptional level and mediates the anti-HBV activity of IFN-γ.
MAPK signaling, especially the ERK1/2 pathway, is activated in response to cytokines and plays a significant role in inhibiting HBV replication [35,36]. Upon activation, ERK1/2 signaling inhibits HBV replication via a range of molecular mechanisms. One of the primary mechanisms by which the MAPK/ERK1/2 pathway hinders HBV replication is through transcriptional regulation of key genes involved in the virus life-cycle. This pathway can modulate the expression of transcription factors and cofactors essential for viral gene transcription, thereby reducing the production of viral RNA and proteins [32,37]. Additionally, activated ERK1/2 affects the post-transcriptional stability of HBV RNA, leading to decreased viral RNA levels in infected cells. Furthermore, MAPK/ERK1/2 pathway activation can trigger a series of downstream events, including the induction of antiviral interferon-stimulated genes (ISGs). Consequently, the combination of these effects on viral gene expression, RNA stability, and host cell factors collectively contributes to host antiviral defense against HBV infection [31,35,38].
HNF4α and HNF1α bind to the HBV enhancer elements, Enh I and Enh II, which are essential for the efficient transcription of HBV genes. By binding to these enhancer sequences, HNF4α and HNF1α promote the transcription of viral genes [39]. HNF4α undergoes regulation through a variety of mechanisms, involving both transcriptional and post-transcriptional processes [40]. Additionally, there is evidence of crosstalk and regulatory interactions between the ERK1/2 pathway and HNF4α/HNF1α [41]. ERK1/2 activation can lead to post-translational modifications of these transcription factors, influencing their activity and DNA-binding properties [42]. For example, the phosphorylation of HNF4 by ERK1/2 inhibits HNF4 transcriptional activity [43,44]. MAPKs are able to downregulate the expression of HNF4α, which leads to less binding between HNF4α and the HBV promoter [40]. Additionally, certain cytokines that activate ERK1/2 can affect the expression of HNF4α and HNF1α, potentially altering their roles in gene regulation [28]. In this study, we identified CIITA as a novel host protein that inhibited HBV transcription by modulating ERK signaling. The data presented in this study demonstrate that CIITA, induced by IFN-γ, decreases the expression of HNF4α and HNF1α by activating the ERK1/2 pathway. Subsequently, this regulation by CIITA reduced enhancer activity, which eventually attenuated HBV transcription (Fig. 8E). The transcriptional regulatory effect of CIITA in other viruses has also been identified. For instance, CIITA blocks the replication of human T-cell leukemia virus type 2 (HTLV-2) by interfering with the function of Tax-2, a major transactivator required for enhancing virus transcription [45].
Several studies have identified the well-developed immune evasion strategies of HBV. In particular, HBx plays a pivotal role in evading host immunity and defense mechanisms [46-48]. In this study, we propose a novel mechanism of HBV immune evasion. Our findings indicate that HBx does not inhibit CIITA expression levels but instead affects CIITA function through direct protein-protein binding (Figs. 6, 7). The interaction between HBx and CIITA increases the protein stability of CIITA but results in its dysfunction. Nevertheless, the mechanism by which HBx inhibits CIITA activity remains elusive and we suggest two possible mechanisms. First, HBx may facilitate the translocation of CIITA, given that the 51–154 amino acid sequence of HBx, which is known to interact with CIITA (Fig. 7), contains a nuclear translocation domain [49]. Secondly, HBx may induce post-transcriptional modifications (PTMs) in CIITA, as it is known for its regulatory influence on various host proteins. Moreover, previous reports have indicated that CIITA is subject to PTMs; specifically, Lys63 ubiquitinated CIITA is concentrated in the cytoplasm [50]. Further research may reveal the exact role of HBx in controlling the anti-HBV activity of CIITA.
As mentioned previously, the classical function of CIITA is to enhance the expression of MHC class II molecules on the surface of antigen-presenting cells (Fig. 8F). Recent studies have demonstrated that CIITA increases the expression of MHC class II molecules in hepatocytes, which act as antigen-presenting cells [51]. Given that HBV-specific CD8+ cytotoxic T cells release IFN-γ, CIITA plays a crucial role in mediating the immune response against HBV by facilitating antigen presentation, in addition to its intracellular anti-HBV action identified here. Similarly, by targeting the transcription of CIITA, Human cytomegalovirus (HCMV) reduces the expression of MHC class II genes, thereby promoting HCMV infection in mature Langerhans cells [52]. Taken together, these findings revealed a novel role of CIITA in regulating HBV transcription and present an immune evasion strategy against HBV (Fig. 8F). The discovery of CIITA as a suppressor of HBV replication opens up new possibilities for the development of therapeutic interventions against HBV infection. Further investigation is needed to elucidate the precise mechanisms of the interplay between CIITA and HBV and to explore the therapeutic potential of targeting CIITA in HBV infection.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Supplement1
Supplement1 Print
Print