Approaches to quantifying hepatitis B virus covalently closed circular DNA
Article information

Abstract
Chronic hepatitis B is a major cause of liver disease worldwide and is currently incurable. Hepatitis B virus (HBV) covalently closed circular (ccc) DNA is a key form of the virus responsible for its persistence and is the transcriptional template for all viral transcripts. The field is focussed on methods to clear HBV cccDNA but this been limited by technical difficulties in its quantification due to: identical sequence to other forms of HBV DNA; low copy number per cell; and high resistance to denaturation by heat, leading to difficulty using polymerase chain reaction or hybridization methods for detection. A number of assays have been developed in order to overcome these hurdles either directly or detecting cccDNA levels indirectly via its transcriptional products. In this review, we summarize the approaches to cccDNA quantification that are currently used, and outline key open questions in the cccDNA biology field which remain to be answered due to the limitations of current methods.
INTRODUCTION
Over 296 million people worldwide currently live with chronic hepatitis B, which causes liver damage, liver cirrhosis and hepatocellular carcinoma (HCC) [1]. Approximately 900,000 people each year die from hepatitis B-related disease. The World Health Organization estimate that only 10.5% of people with chronic hepatitis B virus (HBV) infections are aware of their status and only 16.7% of those diagnosed are on treatment [1].
Current therapies in the form of nucleos(t)ide analogues (NA; such as entecavir, tenofovir disoproxil or tenofovir alafenamide) suppress viral replication and can prevent disease progression and subsequent HCC development [2,3]. However, such treatments need to be taken indefinitely as they do not affect the HBV covalently closed circular (ccc) DNA, the template for all viral transcripts that stably persists in the host cell nucleus [4]. Moreover, the incurability and high risk of liver disease mortality associated with chronic hepatitis B cause multiple psychosocial impacts, e.g., fear, stigma and discrimination [5]. The complete elimination of cccDNA from the liver would represent a cure for chronic hepatitis B, preventing ongoing disease impacts. As such, the induction of cccDNA clearance is a major goal of therapeutic drug development [6,7].
There are many barriers to achieving clearance of cccDNA, one of which is the technical difficulty associated with sensitively, accurately, and precisely quantifying cccDNA levels. This review aims to discuss the importance and challenges of cccDNA quantification, and explore the current molecular methods used for measuring levels in a research environment.
HBV REPLICATION CYCLE
Hepatitis B is caused by infection with the HBV, a blood-borne, hepatotropic virus belonging to the genus Orthohepadnavirus of the Hepadnaviridae family. The virion is composed of a small (3.2 kbp) partially double stranded (ds) DNA genome encapsidated in a viral nucleocapsid that enveloped by a host-derived membrane studded with viral envelope proteins (HBV surface antigen [HBsAg]).
The HBV replication cycle begins with a reversible non-specific interaction between the viral HBsAg on the viral surface to cellular heparan sulphate proteoglycans, particularly glypican 5 [8-10]. The HBsAg on the virus surface then engages in strong irreversible interactions with the cellular sodium taurocholate co-transporting polypeptide (NTCP), expressed only on the basolateral membrane of hepatocytes and conveying liver-specific tropism of the virus. Multiple other co-receptors and host factors also contribute to efficient viral entry. NTCP is localised to the cell membrane through interaction with E-cadherin [11], and complexes with epidermal growth factor receptor in order to mediate viral entry through clathrin-dependent endocytosis [12-14]. Low-density lipoprotein receptor has also recently been implicated in HBV entry, likely by assisting viral attachment via binding to HBV-associated apolipoprotien E [15]. It is probable that additional unknown host factors assist in viral entry. The virus nucleocapsid is released into the cytoplasm and transported to the nucleus along microtubules, where the viral relaxed circular (rc) DNA is released [16].
In the nucleoplasm, rcDNA is repaired by numerous host factors (including TDP2, Pol-K, PCNA, the RFC complex, POLδ, FEN-1 and LIG1 [17-20]) and is converted to cccDNA. cccDNA is complexed with histones [4] and acts as a transcriptional template for the five viral transcripts sufficient for efficient production of virions: 1) the HBx mRNA, translated the viral transcriptional trans-activator (HBV X protein); 2) the PreS and S mRNAs, encoding for HBsAg (large, medium, and small versions); 3) the pregenomic (pg) RNA, which can be translated into the capsid subunit HBV core protein (HBc); 4) the viral polymerase (pol); and 5) the precore RNA, which codes for the secreted protein preCore/core(HBe).
Expression of these transcripts can be affected by acetylation status of histones bound to the cccDNA molecules [21], its chromatin organisation [22], and viral variants that develop over the course of a chronic infection (e.g., mutations in the basal core promoter and precore regions [23-25]).
Transcribed pgRNA and translated pol binds to pgRNA, which triggers encapsidatation by multiple subunits of HBc forming an immature nucleocapsid containing viral RNA [26,27]. HBV pol initiates and carries out reverse transcription of the pgRNA [27-29] through a complicated series of molecular events [30]. Briefly, pol binds to the 5’-epsilon region of the pgRNA and uses this as a template to synthesise a three nucleotide oligonucleotide [31]. This complex translocates to a direct repeat 1 region on the 3’ end of the pgRNA and initiates reverse transcription producing a negative-sense single stranded (ss) DNA strand. During reverse transcription, pol hydrolyses the pgRNA with its ribonuclease H activity 18nt behind the site of reverse transcription [32]. Upon reaching the end of the pgRNA, the pol produces a looped negative sense DNA strand and an 18nt RNA fragment remaining from the pgRNA [33], the latter of which acts as the primer for the synthesis of the positive-sense ssDNA [34]. In 90% of nucleocapsids, the primer translocates to the 5’ end of the ssDNA leading to the synthesis of rcDNA. However in 10% of cases, it remains bound to the 3’ end of the ssDNA, leading to the formation of double stranded linear (dsl) DNA [35]. The mature nucleocapsids (containing HBV DNA) are subsequently enveloped and secreted out of the cell as virions through yet-uncharacterised signals [36].
HBV-infected cells also secrete HBV e antigen (HBeAg) and HBsAg. HBeAg is produced from cccDNA and can be neutralised by host antibodies during robust antiviral response in later phases of infection. On the other hand, HBsAg can be expressed from either cccDNA or integrated HBV DNA (formed when HBV dslDNA is inserted into the host DNA, previously reviewed by Tu et al. [37]). In addition to forming the envelope of virions, HBsAg is also secreted as two forms of non-infectious sub-viral particles: 25 nm diameter spheres, and 22 nm diameter filaments of variable length. Sub-viral particles are produced in excess to virions (by a factor of 1,000- to 100,000-fold) and therefore comprise the major form of HBsAg in the serum [38].
IMPORTANCE OF AND CHALLENGES OF MEASURING cccDNA
Given it is sufficient and necessary for viral replication (and therefore maintaining viral persistence), HBV cccDNA is key to overcoming chronic HBV infections [18]. Elimination of cccDNA is considered a complete cure for HBV infection, and allows for the cessation of therapy without the risk of viral rebound. Understanding of HBV cccDNA biology and the development novel therapeutics inducing its clearance have been hindered by the lack of a simple, specific, sensitive, precise, and high-throughput assay for cccDNA quantification [39]. These technical challenges should each be addressed to improve cccDNA quantification and facilitate ongoing HBV cure research.
Specificity
cccDNA has an identical DNA sequence to all other intracellular and extracellular forms of HBV DNA, including rcDNA, dslDNA, and ssDNA [40]. These different structural forms of HBV DNA present a high risk for false positives. These other forms are present in excess infected cells at ~100–1,000 copies per cell compared to cccDNA at 1–10 copies per infected cell [41].
Sensitivity
In some instances, only small numbers of infected cells are present (e.g., HBeAg-negative patients) or can be isolated from patients (e.g., fine needle aspirates). Given cccDNA can be present at very low levels (<1 cccDNA molecule per 1,000 cells) [42], high sensitivity is necessary for assays to be broadly applicable [42].
Precision/accuracy
Intrinsic to its covalently closed structure, cccDNA resists heat denaturation and thus can be difficult to quantify precisely and accurately by polymerase chain reaction (PCR) or hybridisation methods due to different detection efficiencies [4].
Throughput
Finally, assays with higher throughput are useful for the purposes of drug screening, monitoring patients in trials, and for conducting experiments with multiple conditions.
cccDNA INDIRECT QUANTIFICATION
Indirect quantification of cccDNA can be carried out by measuring the levels of secreted virus gene products (Table 1, Fig. 1). These approaches serve an important role in monitoring levels of HBV patients, as these assays are non-invasive and do not require a traditional core liver biopsy (unlike direct quantification of cccDNA). These viral markers have been linked to disease progression and the development of HCC, suggesting a role for cccDNA levels and clinical outcomes [43]. They have also been used as a highthroughput method to determine cccDNA levels in reporter cell lines and other in vitro systems. In general, however, these indirect cccDNA measures can lack specificity and accuracy for total cccDNA, as they are strongly affected by transcriptional activity of the cccDNA, antiviral therapy, host immune response, and viral factors.
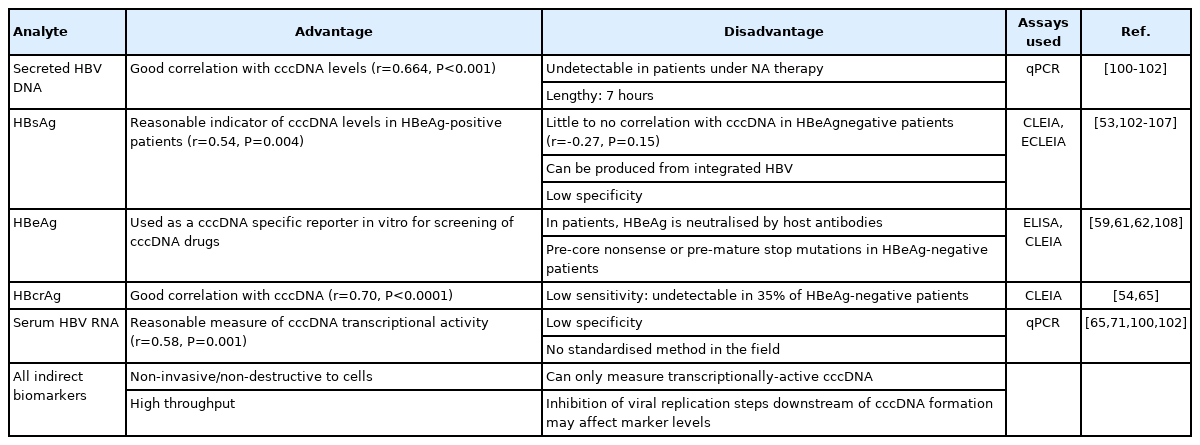
Indirect biomarkers used in cccDNA quantification
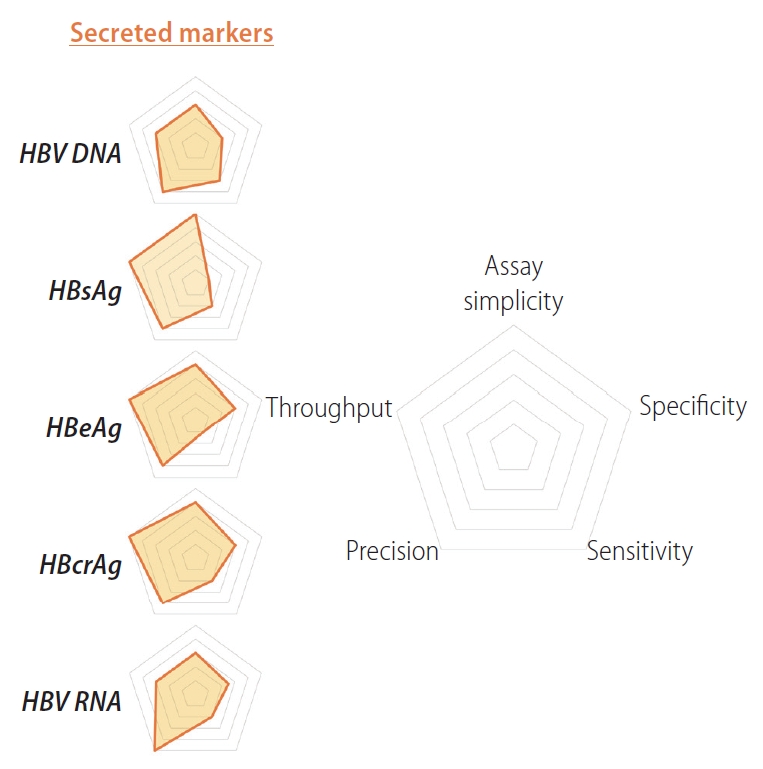
Relative strengths and weaknesses of indirect biomarkers for cccDNA quantification. Radar charts comparing strengths and weaknesses of secreted biomarkers for cccDNA quantification (HBV DNA, HBsAg, HBeAg, HBcrAg and HBV RNA) are plotted on five axes representing: assay simplicity, specificity, sensitivity, precision and throughput. HBV, hepatitis B virus; HBsAg, HBV surface antigen; HBeAg, HBV e antigen; HBcrAg, hepatitis B core related antigen; cccDNA, covalently closed circular DNA.
SECRETED HBV DNA
Secreted HBV DNA (in the form of HBV virions) generally correlates with cccDNA, as virions can only be produced by cells containing cccDNA. However, any factor that inhibits replication steps downstream from cccDNA formation will affect HBV DNA levels. This is seen most obviously with NA therapy, which inhibits reverse transcription and dramatically reduces circulating HBV DNA levels, despite not affecting cccDNA levels significantly [44]. The same is true for experimental agents that affect cccDNA transcription, translation, or secretion. Thus, total HBV DNA is not specific enough for many experimental applications. Secreted HBV DNA is currently detected using real-time quantitative PCR (qPCR) [45].
HBsAg
Serum HBsAg is an important marker for diagnosis of HBV infection. HBsAg is currently detected using automated chemiluminescence enzyme immunoassay (CLEIA) or electrochemiluminescence immunoassay [46], however can also be detected using a standard enzyme-linked immunoassay (ELISA) [47].
HBsAg has been shown to correlate with intrahepatic cccDNA levels in some studies [48-50]. Moreover, serum HBsAg decline during NA therapy has been associated with reduced levels of intrahepatic cccDNA [42]. Other studies, however, have also reported either weak or no correlation with cccDNA levels [51-54].
These disparate findings are potentially be explained by HBsAg originating from the non-replicative integrated form of HBV DNA, making its association with cccDNA unreliable [54]. While the relative level of integration-derived HBsAg is negligible in HBeAg-positive patients (leading to good correlation to cccDNA), a combination of lower cccDNA levels and increased frequency of HBV DNA integrations in later phases [55,56] mean that the majority of HBsAg can be encoded by integrated HBV DNA in HBeAg-negative patients [25,57].
Moreover, it has been reported that HBsAg levels remain relatively static in response to combined peg-interferon (IFN) and lamivudine therapy while cccDNA levels decreased, indicating that there may be a compensatory mechanism related to an increased production of subviral products [58]. Thus, HBsAg cannot be considered an accurate marker for cccDNA levels in HBeAg-negative patients or patients on peg-IFN therapy.
HBeAg
HBeAg is used as a clinical marker for HBV infection. Seroconversion to an anti-HBe state indicates active host immune clearance. During the natural history of the infection, HBeAg levels drop due to both a neutralising antibody response against the antigen and reduction in intrahepatic cccDNA levels as infected cells are cleared by a cell-mediated antiviral immune response. Moreover, under this immune selection, HBV cccDNA variants that cease to express HBeAg can emerge and become fixed in the hepatocyte population. Due to the multiple factors associated with free circulating HBeAg levels, they are poor indicators of cccDNA levels.
In the absence of the complications of an immune system, HBeAg has been used as a reporter for cccDNA in in vitro systems. HepAD38 cells contain an over-length replication-competent HBV DNA transgene, whereby the expression of HBV precore mRNA (and by extension secretion of its translational product HBeAg) is dependent both on cccDNA levels and the activation of the transgene by a tetracycline-controlled promoter [59,60]. To improve the specificity of this system, the HepDE19 cell line was developed so that the HBeAg ORF and its 5’ RNA leader are separated on the transgene, and only become complete following formation of cccDNA [61]. Detection of HBeAg by ELISA therefore would indicate cccDNA levels, useful for high throughput screening of cccDNA targeting drugs [61]. However, a complicating feature came from the high homology between HBeAg and hepatitis B core antigen (HBcAg), the latter of which is secreted in a cccDNA-independent manner by the integrated transgene. To address this, an additional modification to these cells was made: HepBHAe82 were developed to introduce a human influenza hemagglutinin (HA) tag into the precore region, thereby encoding a secreted HA-tagged HBeAg specifically detectable by CLEIA and eliminating interference from HBcAg [62]. HepBHAe82 now can provide the basis of a relatively high-throughput platform to screen for compounds that affect levels of transcriptionally-active cccDNA.
HEPATITIS B CORE RELATED ANTIGEN (HBcrAg)
HBcrAg is a relatively new biomarker detecting the 149 base pair sequence shared between HBcAg, HBeAg and core related protein, p22 [63]. Levels of HBcrAg can be detected using a CLEIA, which is relatively inexpensive and simple compared to PCR for serum HBV DNA.
HBcrAg has the advantage of remaining detectable in many patients with undetectable levels of serum HBV DNA due to NA therapy (106/124 [85%] detectable HBcrAg compared to 36/124 [29%] detectable HBV DNA) [54]. HBcrAg levels have been shown to correlate reasonably well with cccDNA levels [64,65]. However, the standard assay used (Lumipulse G® HBcrAg assay by Fujirebio Europe, Gent, Belgium) has a narrow quantifiable range of 3.0 log10 to 6.8 log10 U/mL, and is therefore somewhat insensitive. In a population of 1,409 untreated HBV patients in the USA, HBcrAg level of many study participants fell below the lower limit of quantification (516/1,409; 36.6%) or had a level higher than the upper limit (318/1,409; 22.6%), which prevented quantification [65]. Samples can be diluted with reagent and retested however, this slows the process down and mitigates the benefit of having a high-throughput system [66].
Considering the main component of HBcrAg is HBeAg, patients in HBeAg-negative phase have lower levels of HBcrAg compared to HBeAg-positive patients [67]. HBcrAg levels are undetectable in many HBeAg-negative patients using current assays (33/94 [35%] undetectable with CLEIA [66]). Correlation with cccDNA levels is dependent on HBeAg levels, with HBcrAg having a good correlation in HBeAg positive patients (r=0.80, P<0.0001), but middling to none in HBeAg-negative patients (r=0.47, P=0.05 in chronic infection patients; r=0.25, P=not significant in chronic hepatitis patients) [66].
SERUM HBV RNA
Serum HBV RNA (in the form of secreted virus particles containing immature nucleocapsids) has been investigated as a potential biomarker for intrahepatic cccDNA transcriptional activity as detected by qPCR [68]. As NA therapy has no effect on cccDNA transcriptional activity, HBV pgRNA-containing virus particles continue to be secreted by hepatocytes with active cccDNA [68] and makes it a superior biomarker compared to serum HBV DNA in patients undergoing treatment, even when HBsAg loss occurs [69].
Serum HBV RNA correlates with intrahepatic HBV-RNA (r=0.73, P<0.001), as well as to the ratio of intrahepatic HBV-RNA to cccDNA (r=0.58, P=0.001). However, HBV RNA is poorly associated with the intrahepatic cccDNA pool (missing 5.62–40.23% of total cccDNA), indicating that some cccDNA forms, e.g., transcriptionally-inactive forms, are undetectable by this method [70,71]. This suggests that serum HBV RNA cannot be used to detect the copy numbers of intrahepatic cccDNA, but can be used to measure cccDNA transcriptional activity.
DRAWBACKS OF INDIRECT METHODS
Despite the usefulness of these surrogate markers, indirect measurements of cccDNA are not perfect replacements for direct measurement. These markers measure transcriptional activity of cccDNA and the rate of conversion from transcriptionally inactive to active forms of cccDNA is still unknown. Therefore, indirect methods cannot give an indication of the actual cccDNA pool size within the liver or determine the potential for viral reactivation.
Another issue which some markers face is their suppression by various therapies that target viral replication downstream of cccDNA formation. Total serum HBV DNA is suppressed by current NA therapies, due to their inhibition of reverse transcription just directly upstream of HBV DNA. In a similar fashion, siRNA-mediated transcriptional suppression silences viral antigens expression, and therefore prevent their detection (despite cccDNA not being affected) [72]. Likewise, core protein allosteric modulators reduce detected HBeAg signals by interfering with assembly or secretion of HBV capsids, but do not affect cccDNA levels [73].
cccDNA DIRECT QUANTIFICATION
Many techniques have been developed to detect and quantify cccDNA itself (including transcriptionally-inactive forms), making them more sensitive and accurate compared to indirect methods (Table 2, Figs. 2, 3). These types of assays however are more invasive in patients or animal models (requiring sampling of the liver) and more destructive in cell culture models (killing the cell to extract DNA or otherwise detect cccDNA). Moreover, direct quantification assays generally take more time to process compared to indirect methods, affecting throughput.

Direct methods of cccDNA quantification

Relative strengths and weaknesses of direct methods for cccDNA quantification. Radar chart comparing relative merits of several approaches to direct cccDNA quantification. Hybridisation methods (green) include Southern blot hybridisation, and fluorescence in situ hybridisation (FISH). qPCR based methods (blue) include qPCR only, or prior enzymatic digestion with plasmid safe DNase (PSD), T5 exonuclease (T5 Exo), and exonuclease 1 and 3 (ExoI/III). Another approach involves serial restriction enzyme digestion and ligation steps prior to qPCR (cccDNA inversion quantitative PCR, cinqPCR). Relative strengths and weaknesses are plotted on the axes as per Figure 1. qPCR, quantitative polymerase chain reaction; cccDNA, covalently closed circular DNA.
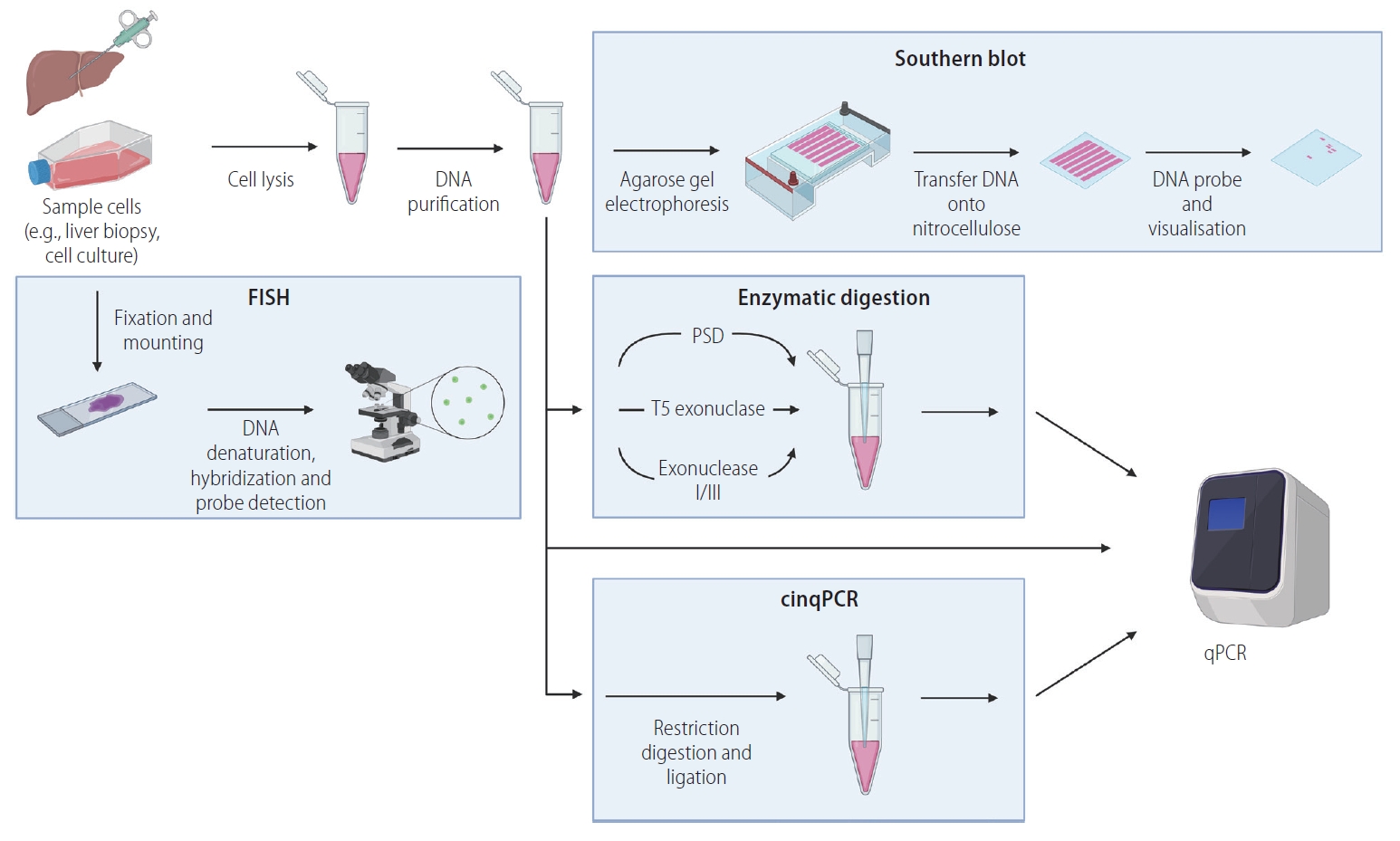
Pathways to directly quantify cccDNA. Several potential technological strategies can be used in combination to directly quantify cccDNA. First, sample enrichment for HBV cccDNA: cells are lysed, and proteins are digested, DNA is purified by precipitation or binding to silica membrane, separating out protein and RNA. Analysis of enriched DNA can then be quantified by Southern blot hybridisation (separation of HBV DNA based on electrophoretic motility through an agarose gel, followed by transfer onto a nitrocellulose membrane and hybridisation with a ssDNA probe). Alternatively, qPCR can be used to quantify cccDNA directly from total DNA, enriched DNA, or after enzymatic digestion (plasmid-safe DNase [PSD], T5 exonuclease, exonuclease I/III, or through cinqPCR). As a third approach, cccDNA can be directly detected by fluorescence in situ hybridisation (FISH): sample cells are fixed and mounted before processing to hybridise DNA with fluorescently marked probes, visualised by immunofluorescent microscopy. Figure created with BioRender.com. cinqPCR, cccDNA inversion quantitative polymerase chain reaction; qPCR, quantitative polymerase chain reaction; cccDNA, covalently closed circular DNA; HBV, hepatitis B virus; ssDNA, single stranded DNA.
DNA SAMPLE ENRICHMENT
Several techniques have been used to enrich for HBV cccDNA (away from other forms of HBV DNA) and improve specificity prior to its detection with the assays described below. Total DNA is generally extracted by lysing the cell and digesting proteins in the solution, followed by separation of DNA from RNA and protein (e.g., by phenol chloroform extraction or binding to silica membranes) [6]. By omitting digestion with proteinase K, protein-bound DNA (including HBV rcDNA) is selectively removed during the extraction process [72].
The Hirt DNA extraction procedure is another method of DNA extraction that selectively isolates low-molecular weight, protein-free (PF) DNA [74,75], including cccDNA and deproteinated rcDNA (an intermediate HBV DNA form produced during the conversion of rcDNA to cccDNA in the nucleus). This method involves lysing cells in a detergent lysis/salt precipitation buffer without proteinase K and extracting the DNA from the supernatant with phenol: chloroform, followed by ethanol precipitation of the DNA. The original Hirt procedure has also been modified to use a silica membrane column for purification of the supernatant [76]. This method, while effective, is laborious and time-consuming even with spin column-based modifications.
Moreover, there is an unknown reduction in cellular DNA extracted for all of these enrichment methods, complicating normalisation and compromising assay precision. Finally, these techniques generally require large amounts of DNA input (microgram range), reducing sensitivity and limiting the source tissues able to be measured.
SOUTHERN BLOT HYBRIDISATION
Southern blot hybridisation is considered the “gold standard” in cccDNA detection as it unambiguously separates out the different forms of HBV DNA out based on electrophoretic motility through an agarose gel [77]. Given the relative low abundance of cccDNA in human infection systems, samples analysed by Southern blot hybridisation are usually first enriched using the Hirt DNA extraction procedure [74,75]. After gel electrophoresis and transfer onto a hybridisation membrane, ssDNA oligonucleotides are used to probe for and visualise HBV DNA.
Southern blot assays are complex, lengthy (~3 days), and insensitive (lower limit of detection ~105 –106 copies of cccDNA) compared to PCR-based assays. Moreover, only a dozen samples can be run on the same gel simultaneously. These drawbacks prevent this method from being used in a high throughput settings, though it is seen as a reliable method for the confirmation of other assays [6].
FLUORESCENT IN SITU HYBRIDISATION (FISH)
FISH-based assays have been used to detect nuclear HBV DNA in cell lines [78]. Using this method, the frequency distribution of nuclear HBV DNA (presumably cccDNA, but may include deproteinated-rcDNA) was shown to average between 8 and 11 molecules per cell in HepAD38 cells. This method of detecting cccDNA can visualise copy numbers of nuclear HBV DNA at a single cell scale, however is time-consuming, labour-intensive, and low-throughput as image analysis requires specialist software. These drawbacks limit its use for methods such as drug screening, but can be a powerful technique for understanding spatial and frequency distribution of HBV cccDNA.
qPCR
Many variations of qPCR have been used to quantify cccDNA, which is simpler, faster and higher throughput compared to hybridisation-based methods [79]. Conventional qPCR was first used by Köck and Schlicht [80] in 1993 using cccDNA-specific primers which span the gap region of rcDNA, preventing rcDNA amplification due to elongation termination at the 5’ end. However, homology between rcDNA and cccDNA-derived products during PCR amplification results in a substantial decrease in specificity, particularly in the presence of excess rcDNA. To resolve this issue, qPCR has been combined with several approaches to enrich for cccDNA over non-cccDNA templates (see “DNA SAMPLE ENRICHMENT” above).
Enzymatic digestion of DNA (either total or Hirt extracted) with exonucleases can be used to remove excess replicative intermediates based on their exposed DNA termini that are absent in cccDNA forms. This approach can be effective, but they risk either: 1) over-digestion and destruction of cccDNA nicked during the extraction procedure; or 2) under-digestion, leading to lower specificity from retention of replicative intermediates. Identifying how long to digest input DNA can be difficult in some instances (e.g., in the presence of high levels of HBV replicative intermediates or when low amounts of total DNA are available). In addition, digestion with these enzymes hydrolyse cellular DNA, and thus complicates precise quantification due to the lack of reliable housekeeping genes for normalisation [73].
Plasmid safe DNase + qPCR
Plasmid safe DNase is an enzyme originally used for removing bacterial chromosomal DNA from plasmid preparations. It preferentially hydrolyses dslDNA, but is less effective against linear and closed circular ssDNAs, and is poorly active against closed circular supercoiled DNA and nicked circular double-stranded DNA [81]. Thus, hydrolysis will retain cccDNA, but also result in incomplete digestion of rcDNA, retaining up to 90% of rcDNA [82-84]. Thus, the specificity of plasmid safe DNase pre-treatment combined with qPCR can be low.
T5 exonuclease digestion + qPCR
T5 exonuclease has been reported to degrade rcDNA for cccDNA purification, showing complete digestion of HBV DNA intermediates, unlike plasmid safe DNase [82]. T5 exonuclease initiates at 5’ termini and degrades both ssDNA and dsDNA in the 5’ to 3’ direction, which allows it to act on nicked dsDNA [82]. Caution is therefore advised for using T5 exonuclease as nicks can be introduced into cccDNA during the DNA extraction process. In addition, care must be taken not to leave T5 exonuclease incubating for too long, as a previous report showed that 16 hours incubation of 5 U T5 exonuclease results in a 60% decrease in supercoiled DNA, while digestion for 60 minutes showed no such effect [82].
Exonuclease I/III digestion + qPCR
A combination of exonuclease I and exonuclease III can be used in order to remove HBV DNA intermediates, with similar efficacy to T5 exonuclease [82]. Both enzymes act in the 3’ to 5’ direction, however exonuclease I specifically degrades ssDNA while exonuclease III is dsDNA specific, preferentially attacking blunt or recessed 3’-protuding termini. These exonucleases lack the single strand endonuclease function of T5 exonuclease, thereby preserving circular ssDNA. This theoretically prevents hydrolysis of cccDNA nicked during the extraction process, increasing accuracy. However, digestion with exonuclease I and exonuclease III also preserves PF-rcDNA intermediates, including a newly discovered HBV DNA form with a covalently closed minus strand with an open plus strand [85,86]. It is currently unknown whether these are true replicative intermediates in formation of cccDNA [85], or if they are simply stable by-products of its generation [86]. Existence of these DNA forms suggests that repair of each DNA strand during cccDNA formation is an independent event [18].
cccDNA INVERSION QUANTITATIVE PCR (cinqPCR)
cinqPCR is a qPCR-based method for detecting cccDNA from total DNA extracts without complete hydrolysis of cellular DNA, allowing normalisation to host genes [73]. Using a series of restriction enzyme digestion and ligation steps [87], HBV cccDNA is converted into an inverted linear form which is efficiently amplified using PCR. Other forms of HBV DNA however are not inverted due to the presence of nicks in a specific region of the viral genome. Together, this means cinqPCR has high accuracy, precision and sensitivity. Our group has now quantified cccDNA in as few as 5 cell equivalents of input DNA and regularly run this assay on a 96-well format. However, this assay is more complicated than the exonuclease-based methods described above and is restricted to detection of the “Galibert” lab strain of HBV, preventing analysis of clinical samples [73].
Due to its high precision, this assay could identify the limited role of HBc on cccDNA levels. We found that wild-type HBV produced similar levels of cccDNA compared to replication-deficient HBV mutant, indicating de novo HBc synthesis was not important for maintaining the cccDNA pool in HBV-infected hepatoma cell lines and primary hepatocytes [88]. Our group had also confirmed the mode of action of different therapeutics (e.g., late treatment with capsid inhibitors does not affect cccDNA levels) [73].
OPEN QUESTIONS REGARDING cccDNA
Many key questions concerning cccDNA biology remain unanswered due to the current limitations of cccDNA quantification. If a highly accurate, precise, and versatile assay were available however, it is possible that cccDNA may be more fully understood and open up novel approaches to remove it thereby curing chronic HBV.
cccDNA HALF-LIFE
The intrinsic lifespan or half-life of cccDNA is of key interest to treating a chronic HBV infection. As new cccDNA formation can be markedly suppressed by current and upcoming therapies, the life-span of cccDNA in the liver determines how effective therapies need to be and how long they must be administered to induce an eventual cure. An ideal cccDNA assay would be able to identify the lifespan of cccDNA by consistent monitoring of cccDNA levels.
Duck HBV models have been combined with mathematical models to estimate cccDNA half-life [89-91], however cccDNA half-life in the human liver is yet to be formally established. Indeed, whether cccDNA decays during therapy or if it even conforms to exponential decay (as implied by the commonly-used term “cccDNA half-life”) has not been shown.
Moreover, partial survival of woodchuck hepatitis virus cccDNA has also been reported. In six out of 10 woodchucks with waning or undetectable surface antigen levels, intrahepatic cccDNA was detected even several years after functional cure [92]. This indicates that either there is low level replication of cccDNA or that cccDNA is highly stable in the liver. Both may be the case: immunosuppression of these woodchucks induced new replication and viral recrudesce [92].
Recent mathematical modelling has indicated that “cccDNA half-life” may vary over the course of infection, depending on host and viral factors. High viral load was associated with a longer and more stable cccDNA half-life of 61 days, while a low viral load was associated with a lower half-life of 26 days [89]. Given this data, it seems unlikely that cccDNA itself has an intrinsic decay rate, but instead its loss is likely mediated by host and viral factors (e.g., cytolytic and non-cytolytic immune responses against HBV-infected cells, cell mitosis, HBV replication and reinfection of hepatocytes). This is consistent with non-dividing in vitro infection models, in which cccDNA levels remained static over 9 weeks [88]. However, this is different to the calculated half-life of duck HBV (3–5 days in culture) [91], suggesting differences between hepadnaviruses of different hosts.
cccDNA DESTABILISATION
Our group and others have shown that cccDNA is highly stable in infected cells not undergoing mitosis [88,91,93]. For example, we found that cccDNA levels were not significantly different between infections with a replication-deficient mutant HBV compared to infections with replication-competent HBV in Huh-7-NTCP, HepG2-NTCP, HepaRG-NTCP and primary human hepatocytes [88]. This strongly suggested that no renewal of cccDNA levels occurs over time within a given infected cell. Thus, direct induction of cccDNA loss (and not inhibition of de novo cccDNA formation) is likely the most efficient approach for complete cure.
Further, chimpanzee studies combined with mathematical modelling have suggested that purely cytopathic methods of cccDNA eradication are unlikely to successfully clear virus [94]. In this model of acute infection, modelling predicted ~11 livers turnovers was required for the observed cccDNA loss, but only ~3 liver turnovers had occurred based on PCNA staining of the liver. To explain the inconsistency, the authors suggested that CD8+ T cells produce IFN-γ within the liver, destabilising cccDNA. An ideal cccDNA assay could possibly identify if or when this process occurred and perhaps even differentiate full-length cccDNA from the destabilised fragments.
Understanding and therapeutically inducing the destabilisation of cccDNA is an intriguing strategy to curing chronic HBV. Interferon-mediated degradation of cccDNA (possibly through regulation of HIF1α) has been reported to induce cccDNA loss [95,96]. Other groups have reported that approaches such as gene editing or epigenetic silencing could also target cccDNA directly and induce its degradation, replication deficiency or transcriptional-silencing [97,98].
MITOTIC LOSS
Mitosis of host cells has been suggested to deplete episomal cccDNA [83], although this has been challenged by other studies indicating the survival of cccDNA in the daughter cells [99]. Reaiche-Miller et al. [99] reported that duck HBV cccDNA could partially survive mitosis based on mathematical models and experimentally derived cccDNA levels in growing duck HBV-infected ducks on NA therapy. Woodchuck models have also shown evidence for some cccDNA molecules being distributed to daughter cells [93]. In contrast, human liver chimeric mice seeded with HBV-infected cells have shown dramatic reductions of cccDNA during cell mitosis, consistent with complete cccDNA loss [83]. Such disparate findings in different animal models must be resolved in order to answer the question on the fate of cccDNA following mitosis of the host cell. A highly-sensitive and -specific cccDNA assay could quantify cccDNA on a single-cell level to directly determine if loss, unequal partitioning, or survival of cccDNA occurs in daughter cells upon mitosis.
CONCLUSION/SUMMARY
HBV cccDNA is central to the maintenance of chronic infection and key questions in cccDNA biology still remain unanswered. To improve our knowledge and facilitate HBV cure research, cccDNA must be accurately quantified and monitored over time, either through indirect biomarkers or via direct measurement. All current methodologies have strengths and weaknesses, with indirect measures being generally faster and less invasive but less accurate, and direct methods slower and more precise. Open questions on cccDNA biology will remain until more advanced methods of quantification can be developed.
Notes
Authors’ contributions
Both authors contributed equally to the writing of this manuscript.
Conflicts of Interest
The authors have no conflicts to disclose.
Acknowledgements
T.T. is funded by Australian Centre for HIV and Hepatitis grant (annual grant) and the Australian National Health and Medical Research Council (Ideas Grant GNT2002565).
Abbreviations
cccDNA
covalently closed circular DNA
cinqPCR
cccDNA inversion quantitative PCR
CLEIA
chemiluminescence enzyme immunoassay
dsDNA
double stranded DNA
dslDNA
double stranded linear DNA
ELISA
enzyme-linked immunoassay
FISH
fluorescence in situ hybridisation
HA
hemagglutinin
HBc
HBV core protein
HBcAg
hepatitis B core antigen
HBcrAg
hepatitis B core related antigen
HBeAg
HBV e antigen
HBsAg
HBV surface antigen
HBV
hepatitis B virus
HCC
hepatocellular carcinoma
IFN
interferon
NA
nucleos(t)ide analogues
NTCP
sodium taurocholate cotransporting polypeptide
PCR
polymerase chain reaction
PF
protein-free
pgRNA
pregenomic RNA
pol
viral polymerase
qPCR
quantitative PCR
rcDNA
relaxed circular DNA
ssDNA
single stranded DNA