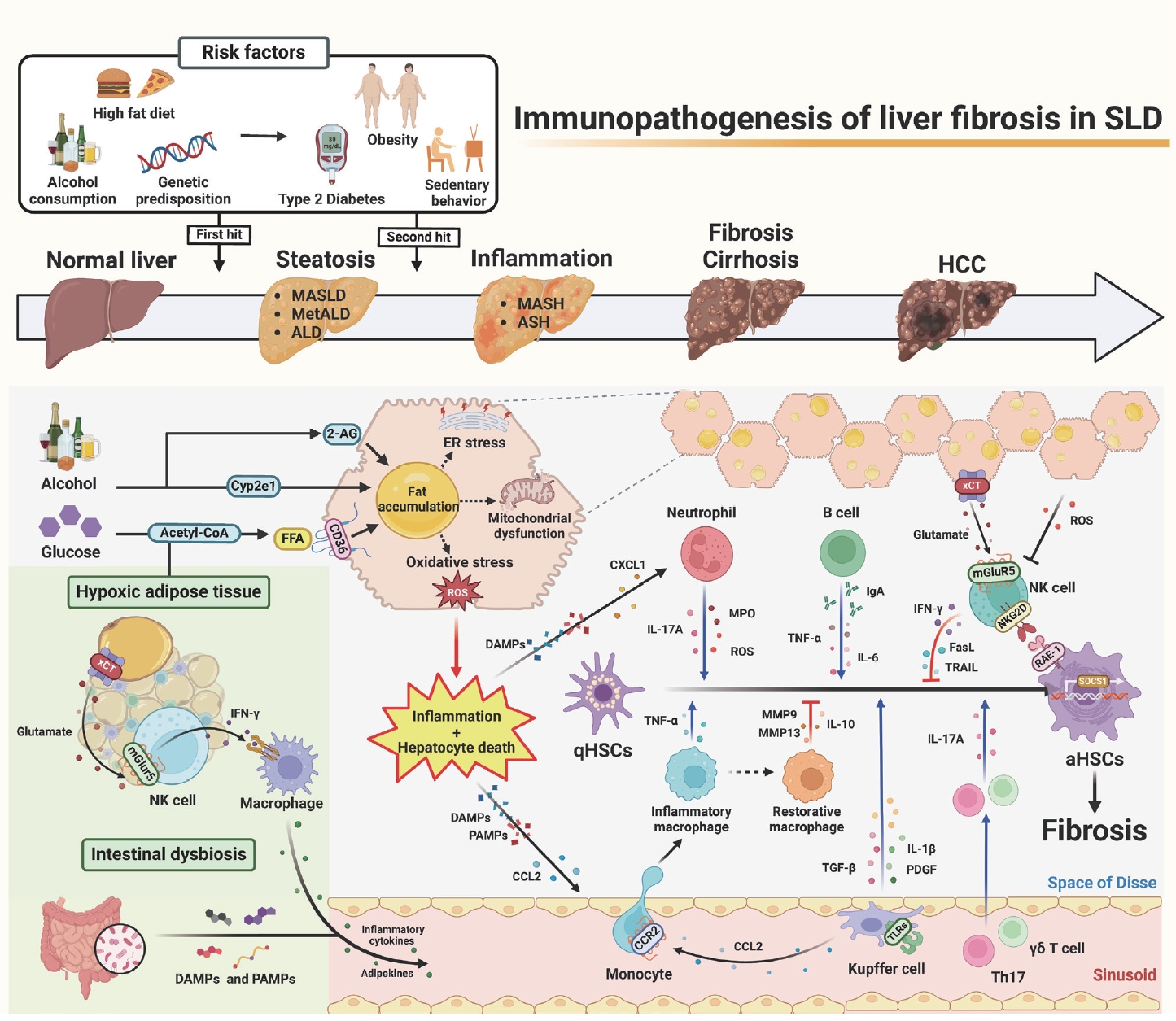
Recently, a new term, steatotic liver disease (SLD), has been introduced to the field of hepatology to encompass the various etiologies of steatosis [1,2]. Hepatic steatosis, having an excessive fat accumulation, has been considered a key factor in the progression of serious liver diseases such as hepatic inflammation, fibrosis/cirrhosis, and hepatocellular carcinoma [2-4]. It could be initiated by an overload of nutrition (e.g., glucose and free fatty acids), intoxicants (e.g., alcohol and endocannabinoid-mediated metabolic dysfunction), or genetic factors [3-5]. Interestingly, lines of evidence have suggested that beyond its role as a metabolic organ, the liver is considered an immunological and neurological organ due to its diverse metabolic functions, enriched immune cells (e.g., innate and adaptive immune cells), and production and release of hepatotransmitters such as glutamate or 2-arachidonoylglycerol (2-AG) [4-6]. So, these factors might affect the progression of SLD, from simple steatosis to steatohepatitis and fibrosis, by regulating the activation of hepatic stellate cells (HSCs).
It has been reported that inflammation and subsequent liver fibrosis occur due to various immune-mediated activations of HSCs through eating habit-mediated metabolic dysfunction (e.g., obesity, type 2 diabetes, or chronic alcohol consumption), sedentary behavior, and genetic predisposition [4,5]. Steatohepatitis may be triggered by prolonged multiple stresses in fat-storing hepatocytes, such as endoplasmic reticulum (ER) stress, mitochondria dysfunction, and oxidative stress, including alcohol-induced reactive oxygen species (ROS) and lipotoxicity [5,6]. These cellular stresses, combined with diverse harmful factors, such as inflammatory cytokines, adipokines, short-chain fatty acids, and pathogen- or damage-associated molecular patterns (PAMPs or DAMPs) originated from adipose tissue (AT) and the intestine, may cause hepatocyte death and inflammation [7]. Recently, an interesting study suggested glutamate-mediated cross-talk between AT and liver, in which hypoxic adipocytes excreted glutamate through the xCT transporter and the resulting production of interferon-╬│ (IFN-╬│) in natural killer (NK) cells by metabotropic glutamate receptor 5 (mGluR5), subsequently leading to the augmentation of adipose macrophage activation and steatohepatitis in obese mice and patients [8].
Through these cellular responses and organ cross-talks, inflammatory immune cells migrate nearby damaged hepatocytes and stimulate the transformation of quiescent HSCs (qHSCs) into activated HSCs (aHSCs) by producing various mediators, including cytokines, chemokines, and extracellular vesicles. Injured fat-storing hepatocytes produce chemokines, such as C-C motif chemokine 2 (CCL2) and CXC motif chemokine ligand 1 (CXCL1), to recruit pro-inflammatory macrophages and neutrophils, respectively. After migration, macrophages, neutrophils, and resident Kupffer cells are stimulated by DAMPs (e.g., ATP, mitochondrial DNA, or double-stranded RNA), PAMPs (e.g., LPS, CpG DNA, Flagellin), or cytokines through specific receptors, including P2X purinoceptor 7 and toll-like receptors (e.g., TLR2, TLR3, TLR4, TLR5, and TLR9) [7]. Then, the production of tumor necrosis factor (TNF)-╬▒, interleukin (IL)-1╬▓, platelet derived growth factor (PDGF), IL-17A, myeloperoxidase (MPO), transforming growth factor (TGF)-╬▓1, and ROS by these cells leads to the activation of qHSCs [9,10]. Moreover, several types of lymphocytes, such as Th17 cells, ╬│╬┤T cells, and B cells, augment HSC activation by producing IL-17, TNF-╬▒, and IL-6 [9,11,12].
In contrast, several types of cells, such as NK cells and restorative macrophages, can kill aHSCs or suppress HSC activation by producing death ligands, including TNF-related apoptosis-inducing ligand (TRAIL) and Fas ligand (FasL), IFN-╬│, anti-inflammatory IL-10, and matrix metalloproteinases (MMPs) [9,13]. Once activated HSCs express retinoic acid early inducible 1 (RAE-1), a specific ligand for NKG2D, NK cells specifically produce IFN-╬│, TRAIL, or FASL through the NKG2D-RAE-1 interaction [13]. In addition, Ly6Clow F4/80+ CD11b+ macrophages can induce fibrosis resolution by producing MMPs or Gr1+ CD11b+ bone marrow cells, which inhibit inflammation by IL-10 production in the early stage of liver fibrosis [9,14]. However, prolonged injuries (e.g., chronic alcohol consumption) cause HSCs to have the ability to avoid this cytotoxicity by increasing TGF-╬▓ production and suppressor of cell signaling 1 (SOCS1) expression in aHSCs [5,15]. Moreover, ROS might deplete NK subpopulations or suppress NK cytotoxicity in advanced liver fibrosis [16,17]. A recent study suggested an interesting solution for how to overcome HSC evasion against NK cytotoxicity, in which mGluR5 activation in NK cells by hepatic glutamate, a hepatotransmitter, further enhanced NK cytotoxicity to aHSCs, resulting in the attenuation of liver fibrosis [17]. Accordingly, the delivery of mGluR5-stimulated NK cells through the tail vein improved liver fibrosis in mice [17], suggesting a therapeutic intervention of liver fibrosis in patients.
In the past, SLD was somewhat neglected, but as viral hepatitis has recently been overcome, the possibility of SLD progression to serious chronic liver disease has emerged. Despite pro-inflammatory immune responses for SLD progression to liver fibrosis, it is noteworthy that certain immune cells (e.g., NK cells or restorative macrophages) may have strong anti-fibrotic functions. In particular, new therapeutics can be developed by carefully looking at intrahepatic neurological signaling (e.g., 2-AG production in HSCs and mGluR5 activation in NK cells). Therefore, precise molecular pathways related to the immunopathogenesis of liver fibrosis should be extensively investigated. Notably, further research will be needed to determine how the neuro-metabo-immune axis, still minimally investigated, influences SLD and liver fibrosis.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print