INTRODUCTION
Porphyria is a class of disorders of porphyrin and porphyrin-precursor metabolism resulting from aberrations in the control of the heme biosynthetic pathway. Erythropoietic protoporphyria (EPP) is an autosomal dominant/recessive porphyric disorder characterized by the accumulation of protoporphyrin in blood, erythrocytes and tissues resulting from a deficiency in the enzyme ferrochelatase.1-3 The incidence of EPP in the population ranges from 1:75,000 to 1:200,000.4 EPP manifests clinically as immediate painful photosensitivity of the skin, with stinging, prickling and burning of exposed areas being the initial symptoms experienced during or shortly after exposure to the sun.4 Long sunlight exposure may result in erythema, edema, petechia and/or bulla formation.4
Because protoporphyrin is a lipophilic molecule that is excreted by the liver, EPP patients are at risk of cholelithiasis with obstructive episodes, and a small minority of EPP patients develop chronic liver disease due to pigment-loading of hepatocytes and bile canalicular sludging.4,5 Inadequate biliary excretion of protoporphyrin has toxic effects on hepatobiliary structure and function. Exposure of cultured hepatocytes to protoporphyrin has been found to inhibit cell metabolism and to increase cellular fragility.6-8 The resultant bile may damage the bile duct epithelium, resulting in biliary fibrosis.4,5 Patients with severe EPP and hepatic involvement frequently undergo hepatic or bone marrow transplantation.9-12
We describe here a patient with acute-on-chronic liver failure from EPP who was treated by urgent liver transplantation (LT), with special attention to specific shielding from light exposure.
CASE REPORT
The patient, a 31-year old male with photosensitivity since childhood, had a family history on his mother's side of photosensitivity of unknown cause. Two years earlier, he had been diagnosed with porphyria and liver cirrhosis by skin and liver biopsy. Fatigue and abdominal distention continued for 10 months despite medical treatment. Due to a lack of improvement of liver cirrhosis-related symptoms, he was enrolled on the waiting list for deceased-donor LT. While on the waiting list, however, he experienced two episodes of hepatic encephalopathy. A genetic study, using polymerase chain reaction followed by sequencing, confirmed the diagnosis of EPP and the presence of a FECH gene mutation (18q21.3). His symptoms and laboratory profiles aggravated rapidly: total bilirubin 25.4 mg/dL, albumin 2.2 g/dL, creatinine 2.5 mg/dL, and prothrombin time 2.71 INR. He finally fell into acute-on-chronic liver failure with hepatorenal syndrome, with a model for end-stage liver disease (MELD) score of 39. We then searched for a living donor for LT, but his younger brother had the same genetic mutation, making the latter ineligible for donation. The patient was given priority allocation for deceased donor organs. His condition worsened as he became semicomatose and his total bilirubin concentration increased to 45.6 mg/dL. Continuous renal replacement therapy was promptly started. After a wait of 3 days, he underwent urgent deceased-donor LT.
Just after laparotomy, his entire viscera were protected from light in the operating room; this was because exposure to intense light is a risk factor for bowel perforation in patients with EPP. We therefore used low-powered light emitting diode (LED) light sources and a wavelength-adjusted filter film. The patient underwent classical standard transplantation using active venovenous bypass. A rubber T-tube was inserted to the biliary anastomosis.
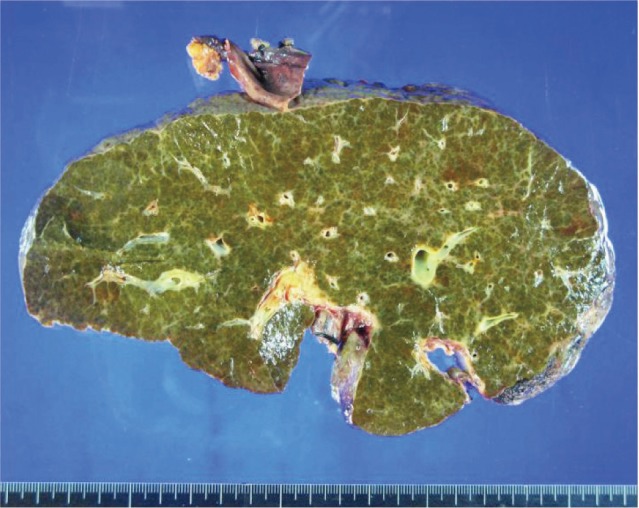
The patient's native liver weighed 1427 g; the parenchyma was colored dark and brownish green and had multiple cirrhotic nodes (Fig. 1). Microscopically, we observed regenerating submassive hepatic necrosis with accumulation of dark-brown pigments and severe cholestasis (Fig. 2A). Polarizing microscopy showed that these pigments had yellow and red birefringence in a Maltese cross pattern (Fig. 2B). All of these findings supported the diagnosis of EPP.5,6,9,13
Posttransplant recovery was slow but the patient improved progressively. He recovered consciousness, but tracheostomy was performed on posttransplant day 18 due to weak respiratory muscle power. After weaning from the ventilator, the tracheostomy cannula was removed on day 42. He was kept on continuous renal replacement therapy and conventional hemodialysis for 2 months until his renal function recovered. Total porphyrin concentrations in his blood and urine were measured serially, disappearing on qualitative analysis after recovery from LT. His immunosuppressive regimen included cyclosporine (target trough concentration, 120-150 ng/mL for the first 3 months) mycophenolate (target trough concentration, 1-3 ng/mL for the first 3 months) and methylprednisolone. The safety of all drugs was checked using the drug database for acute porphyria (http://www.drugs-porphyria.org), to avoid the use of porphyrogenic agents.5
He was hospitalized for 3 months after transplantation due to the requirement for rehabilitative treatment, enabling him to stand and walk. At present, 24 months after LT, the patient is doing well, with no complications.
DISCUSSION
EPP is a rare disease, with an incidence in the general population ranging from 1:75,000 to 1:200,000.3-5 The first LT for EPP-related liver failure was reported in 1979.9 In Korea, EPP is very rare, and this patient is the first in Korea with EPP to undergo LT. Of 2500 adult LTs performed at our center, this patient was the only one with EPP.
EPP results from abnormalities in ferrochelatase activity.2,3,9,14,15 This enzyme converts protoporphyrin to heme in the final step of the heme biosynthetic pathway. In the absence of ferrochelatase activity, protoporphyrin accumulates in blood, erythrocytes, bone marrow and other tissues. As protoporphyrin is excreted by the liver, excess protoporphyrin results in high concentrations of protoporphyrin within hepatocytes, producing crystalline deposits and additional cell injury. Altered bile composition and inadequate biliary excretion of protoporphyrin has toxic effects on hepatobiliary structure and function.4,8,9 Bile formation is impaired and bile duct epithelium is damaged, resulting in biliary fibrosis.4 Thus, patients with EPP are at risk of cholelithiasis with obstructive episodes, and a small proportion develops chronic liver disease. Progression of protoporphyric hepatic deterioration leads to splenomegaly, splenic sequestration of erythrocytes with hemolysis and protoporphyrin generation, ending in fulminant hepatic failure.5,8
EPP is an inherited disorder with both recessive and dominant patterns of inheritance.5,13 Most patients with EPP have one allele with a FECH mutation and the low-expression allele.3,5 The FECH gene was cloned and sequenced in 1990 and localized to the long arm of chromosome 18 (18q21.3).2,3,13,16
The patient in the present study had the FECH gene mutation c.386del (p.Gly129AspfsX16), which had not been reported previously. His younger brother had the same mutation, but he did not have symptoms of EPP. The latter is therefore an asymptomatic gene carrier and thus could not be a liver donor. Because he has 50% FECH activity and a normal phenotype, the pedigrees of this patient and his brother should be evaluated extensively.
Bowel protection is important during surgery in porphyria patients because laparotomy itself may carry risks of bowel perforation.6,7,9 The visceral organs can be damaged by phototoxic reactions of astral lamps and the operating room lights, resulting in third-degree burns and delayed bowel perforation. We therefore used low-powered LED lights and a wavelength-adjusted filter film. Since blue, violet and ultraviolet-A (400-410 nm wavelength) light excite protoporphyrin, they must be blocked effectively in the operating room.7 LED light releases less ultraviolet radiation than the lights usually used in the operating room. The LED lights were covered by a wavelength-adjusted yellow film that could block blue, violet and ultraviolet light. Although the power of the light source was decreased and operating field was much darker than during usual laparotomy, the LT operation was not compromised because it was a routine surgical procedure. Even after LT, we lowered the intensity of ambient light during intensive care due to possible photosensitivity. After full physical recovery, no light barrier was applied except for direct exposure to sunlight. The intraoperative light protection is very similar to the patients undergone photodynamic therapy, but the period for posttransplant protection was relatively shorter in EPP patients than in patients receiving photodynamic therapy.
The immunosuppressive regimen for this patient consisted of the triple combination of cyclosporine, mycophenolate and methylprednisolone. Cyclosporine is a safe immunosuppressant for patients with porphyria, according to the drug database for acute porphyria.7 In addition, cyclosporine has been reported to inhibit the mitochondrial permeability transition of hepatocytes that is induced by protoporphyrin IX, whereas tacrolimus does not.15 Reduced hepatotoxicity may enhance biliary excretion and account for the marked decrease in plasma protoporphyrin concentrations. Cyclosporine has been suggested as the calcineurin inhibitor of choice and may be beneficial in the treatment of nontransplant protoporphyrin hepatopathy.15
Several reports have assessed survival after EPP-related LT. For example, the overall 1-, 5-, and 10-year patient and graft survival rates in 20 EPP patients who underwent LT in the United States between 1979 and 2004 were 85%, 69%, and 47%, respectively.9 In pediatric patients, the 1-, 5-, and 10-year patient and graft survival rates were 100%, 75%, and 50%, respectively.9 Recurrent EPP liver disease occurred in 11 (65%) of 17 patients who survived more than 2 months after LT. Recurrent EPP liver disease occurred as early as 8 months after LT, and three patients required retransplantation, at 1.8, 12.6, and 14.5 years, respectively. Three additional patients who died with recurrent EPP liver disease had extensive protoporphyrin deposits and bridging fibrosis or cirrhosis on liver biopsy. Recently, European multi-center study, including UK data, showed the overall patient survival rates of 77% at 1 year and 66% at 5 and 10 years in 31 patients.17,18
Although the effective treatment of liver failure caused by EPP is LT,14 it does not ameliorate the underlying protoporphyrin overproduction in the native bone marrow, thus leaving the recipient at risk for recurrent disease.9 Bone marrow produces 80% of protoporphyrin IX and an optimal therapy for EPP would correct the defect in heme synthesis.5,9-12 Sequential liver and bone marrow transplantation may be used to treat patients with EPP.10,12 Bone marrow transplantation can alter the genotype of bone marrow, resulting in phenotypic reversal, thus protecting patients from future protoporphyric liver disease.12,19 Bone marrow transplantation may be indicated for EPP patients without advanced liver fibrosis after reversal of liver failure, for older patients with recurrent graft disease after LT, for younger patients after LT, and for patients with progressive liver disease. The patient described here may undergo future bone marrow transplantation because he is young and considered at high risk for EPP-related liver disease.12 We have performed bone marrow transplantation in a LT recipient to treat hematologic malignancy. Owing to his familial history, an extensive search for an appropriate donor would be necessary. Future prevention and treatment of liver disease in this patient may include erythrocyte transfusion, plasmapheresis, ursodeoxycholic acid, cholestyramine, charcoal and tocopherol, as well as bone marrow transplantation.3,5,9-12,20

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print